

- Department of Neurosurgery and Medical Informatics, Nagoya City University Graduate School of Medical Sciences, Nagoya, Aichi, Japan.
- Department of Neurosurgery, Nagoya City University Graduate School of Medical Sciences, Nagoya, Aichi, Japan.
Correspondence Address:
Hiroyuki Katano
Department of Neurosurgery, Nagoya City University Graduate School of Medical Sciences, Nagoya, Aichi, Japan.
DOI:10.25259/SNI_387_2020
Copyright: © 2020 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Hiroyuki Katano1, Yusuke Nishikawa2, Hiroshi Yamada2, Takashi Iwata2, Mitsuhito Mase2. Profile of genetic variations in severely calcified carotid plaques by whole-exome sequencing. 12-Sep-2020;11:286
How to cite this URL: Hiroyuki Katano1, Yusuke Nishikawa2, Hiroshi Yamada2, Takashi Iwata2, Mitsuhito Mase2. Profile of genetic variations in severely calcified carotid plaques by whole-exome sequencing. 12-Sep-2020;11:286. Available from: https://surgicalneurologyint.com/?post_type=surgicalint_articles&p=10254
Date of Submission
26-Jun-2020
Date of Acceptance
19-Aug-2020
Date of Web Publication
12-Sep-2020
Abstract
Background: The precise mechanisms of carotid calcification and its clinical significance have not been established.
Methods: We classified ten plaques from carotid endarterectomy patients into high- and low-calcified plaques based on the Agatston calcium scores. We performed whole-exome sequencing for genetic profiles with single nucleotide variations (SNVs), insertions, and deletions. Bioinformatic data mining was then conducted to disclose specific gene variations to either high- or low-calcified carotid plaques.
Results: In the carotid plaques, G:C>A:T/C:G>T:A transitions as SNVs, insT after C/insC after A as insertions, and delA after G/delT after C as deletions were most frequently observed, but no significant difference was observed between the high- and low-calcified plaque groups in their proportion of base-pair substitution types. In the bioinformatic analysis, SNVs of ATP binding cassette subfamily C member 6 (ADCC6) were more commonly found in high-calcified plaques and SNVs of KLKB1 were more commonly found in low-calcified plaques compared to the other group. No new genetic variants related to calcification or atherosclerosis among those not registered in dbSNP was detected.
Conclusion: Our findings clarified the features of base-pair substitutions in carotid plaques, showing no relation to calcification. However, genetic variants in ADCC6 relating to vascular calcification for high-calcified plaques, and in KLKB1 encoding kallikrein associated with vascular regulation of atherosclerosis for low-calcified plaques were more specifically extracted. These results contribute to a better understanding of the genetic basis of molecular activity and calcium formation in carotid plaques.
Keywords: Calcification, Carotid endarterectomy, Carotid plaque, Single nucleotide variation, Whole exome sequencing
INTRODUCTION
Calcification in carotid plaques is often encountered in patients with asymptomatic carotid stenosis, and this calcification has been thought to be stable and hardly evoke symptoms compared to so- called “soft” vulnerable plaques.[
Single nucleotide polymorphisms (SNPs) are naturally occurring variants that affect a single nucleotide and are present in >1% of a population. SNPs are present in the germline of an individual, whereas single nucleotide variations (SNVs) are single-base differences among individuals, and their percentage in a population is not a factor; SNVs are also observed in somatic cells. Although atherosclerotic diseases such as cerebrovascular disease (CVD) and coronary artery disease (CAD) are multifactorial disorders, and information about the genetic associations in their pathogeneses is limited, associated SNPs have been reported in trials seeking influential rare variants or genetic biomarkers[
In the present study, we used an approach that differed from those used in the above-cited conventional studies of CVD that sought to identify SNPs using large cohorts and peripheral blood sampling. We attempted to determine the differences in variations of genes by whole-exome sequencing (WES), according to the presence and amount of calcification in carotid plaque specimens that were obtained during carotid endarterectomy (CEA) for patients with carotid stenosis. We applied bioinformatic data mining to disclose the specific susceptibility gene variations associated with high- or low-calcified plaques to gain a better understanding of the genetic basis for calcification in carotid plaques.
MATERIALS AND METHODS
Patients and specimens
Carotid plaques from ten endarterectomy patients were investigated [

All of the patients enrolled in this study received a detailed explanation of the nature of the project, and all gave written informed consent before undergoing the CEA. Approval from the local ethical committee for human genetic research was also obtained.
The patients underwent preoperative multi-detector computed tomography angiography (MDCTA) that identified the plaque location and the degree of stenosis. The MDCTA was performed with a 64-detector row CT scanner (SOMATOM Definition: Siemens Medical Solutions, Forchheim, Germany). An evaluation of the calcification of the plaques using the Agatston calcium score was also performed with non-contrast- enhanced CT obtained before the contrast media injection for MDCTA, as described elsewhere.[
Briefly, the calcification of each carotid plaque was quantified using specialized software run on a workstation (Ziostation ver. 1.17; Amin, Tokyo) with the preoperative MDCTA data. The Agatston calcium score was calculated as the product of the areas of calcified lesions and the weighted signal intensity scalars, dependent on the maximal Hounsfield unit (HU) value within the lesion (scalar = 1 if 130–199 HU, 2 if 200– 299, 3 if 300–399, and 4 if ≥400).
All carotid plaques were obtained during a CEA, and specimens containing portions consisting of the most severe stenosis were harvested. This enabled the sampling of tissues with high calcification for the plaques with high calcium scores and the sampling of tissues with less calcification for the plaques with low calcium scores. All samples were stored at −80°C and thawed only once.
WES
Genomic DNA was extracted from fresh frozen carotid plaques using a Nucleospin Tissue Kit (Macherey Nagel, Düren, Germany) following the manufacturer’s instructions. DNA was qualified using a NanoDrop and agarose gel electrophoresis. DNA libraries were prepared and exome enrichment was performed by SureSelect XT Human All Exon V6 (Agilent Technologies, Santa Clara, CA). The 100-bp end sequencing of exomes was carried out on the HiSeq2500 genome analyzer platform (Illumina, San Diego, CA). The sequencing raw reads were mapped to the UCSC hg19/GRCh37 reference human genome by the Burrows- Wheeler Aligner-MEM ver. 0.7.15. Duplicate marking and base quality recalibration were performed using Picard ver. 2.2.4 (
Bioinformatics data mining
All detected variants were filtered to exclude low-quality variants with the use of GATK by choosing those with “PASS” in the FILTER field. The effects of alterations of amino acids and frameshift variants on the protein structure and function were predicted as the Annotation Impact of “HIGH” or ‘MODERATE’ by SnpEff ver. 4.2.[
Among the gene variations that satisfied all of the above- noted conditions, we compared the high- and low-calcified plaque groups and chose the variations with the mean allele frequency (AF) and a fold change >2 compared to the other group. We then compared all 25 individual sets of the two groups (one of five high-calcified plaques vs. one of five low- calcified plaques) and selected the unique gene variations detected in only the high- or low-calcified plaque group in >30% of the comparison sets. In both comparisons, we excluded the gene variations with 0.4≤ (mean) AF ≤0.6 that were suspected to have higher possibilities of germline origin. The gene variations that were detected in both groups and in individual comparisons were extracted as the final results.
To identify unknown novel gene variations in the calcified carotid plaques, we selected genes that were not included in the database SNPs (dbSNP) (
We performed an enrichment analysis for biological pathways of genes using the functional annotation clustering algorithm on the database for annotation, visualization, and integrated discovery (DAVID ver. 6.8,
Statistical analyses
All statistical evaluations were performed with the statistical software SPSS ver. 22.0 (SAS, Cary, NC, USA) and StatMate III (ATMS, Tokyo), and all results are presented as mean ± SD values. The Mann–Whitney U-test and the Chi- square test with Yates’ correction were used to compare the demographic data between the patients with high- and low-calcified plaques. The between-group differences in the proportions of variables were evaluated with Spearman’s rank correlation coefficient, and those between individual specimens were evaluated with Pearson’s correlation coefficient. P < 0.05 was considered significant.
RESULTS
Genetic variants detected by the WES
About 99.6–99.7% of the sequence reads were mapped to the UCSC hg19 reference human genome. The coverage rate was 83.1–91.2% by ≥20 unique reads, and 72.4–74.0% of the target regions were covered. Of all 13,783,666 detected variants, SNVs, insertions, and deletions accounted for 88.0%, 5.3%, and 6.8%, respectively. The proportions of the identified principal variants in the targeted exome region are shown in [
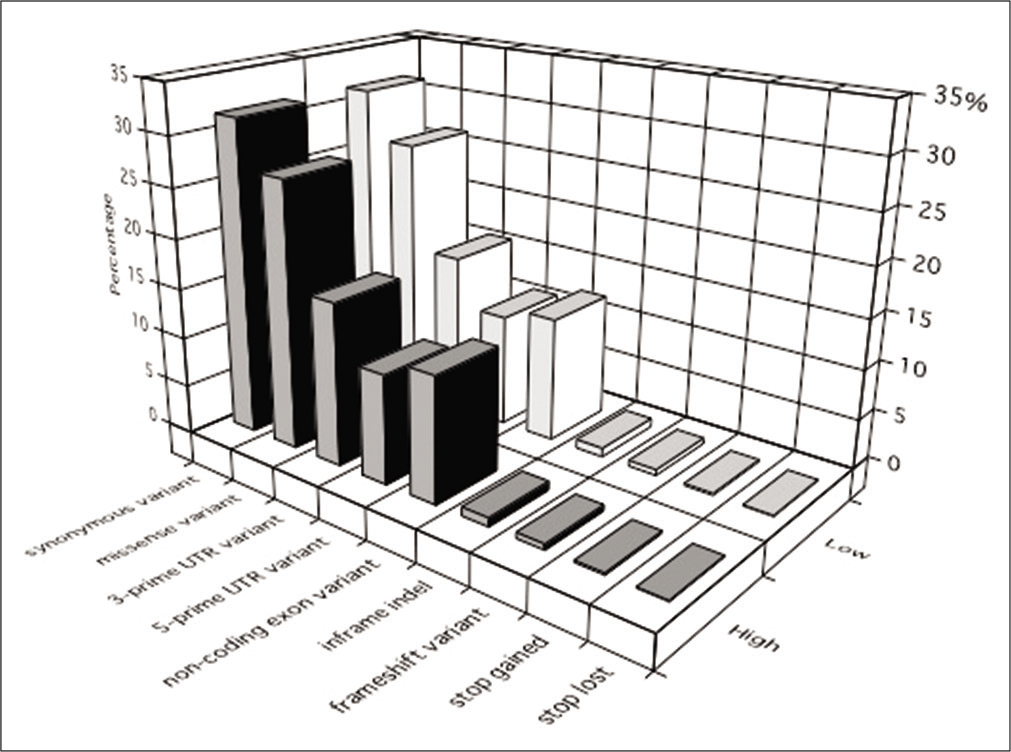
Figure 1:
The proportions of the identified principal variants in the targeted exome region. Synonymous and missense variants represented the most common types of variants in all specimens. Similar proportions of variants were observed in the high- and low- calcified plaque groups. High: high-calcified plaques, Low: low- calcified plaques.
The percentages of SNVs (r = 0.94–0.99, P < 0.001), insertions (r = 0.55–0.93, P < 0.001), and deletions (r = 0.72– 0.96, P < 0.01 or P < 0.001) in individual plaques shown in [
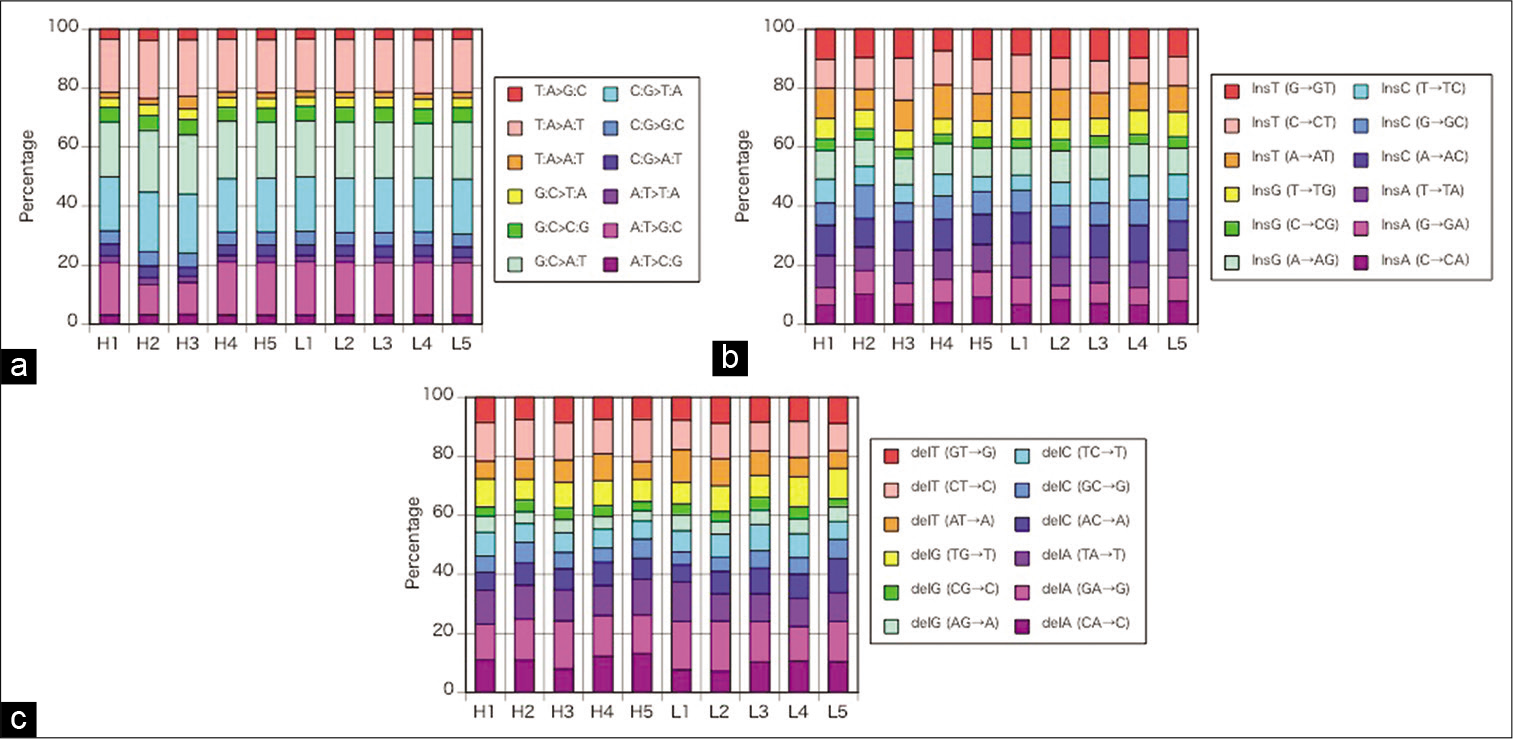
Figure 2:
(a) The percentages of single nucleotide changes in individual plaques demonstrated no remarkable difference in each specimen for base-pair substitution. (b) The percentages of insertions in individual plaques demonstrated no remarkable difference in each specimen for base-pair substitution. (c) The percentages of deletions in individual plaques demonstrated no remarkable difference in each specimen for base-pair substitution. H1–5: high-calcified plaques, L1–5: low-calcified plaques.
Regarding the SNVs, G:C>A:T and C:G>T:A transitions were the most frequent followed by T:A>A:T transversion and A:T>G:C transition [ρ = 1, P < 0.01,
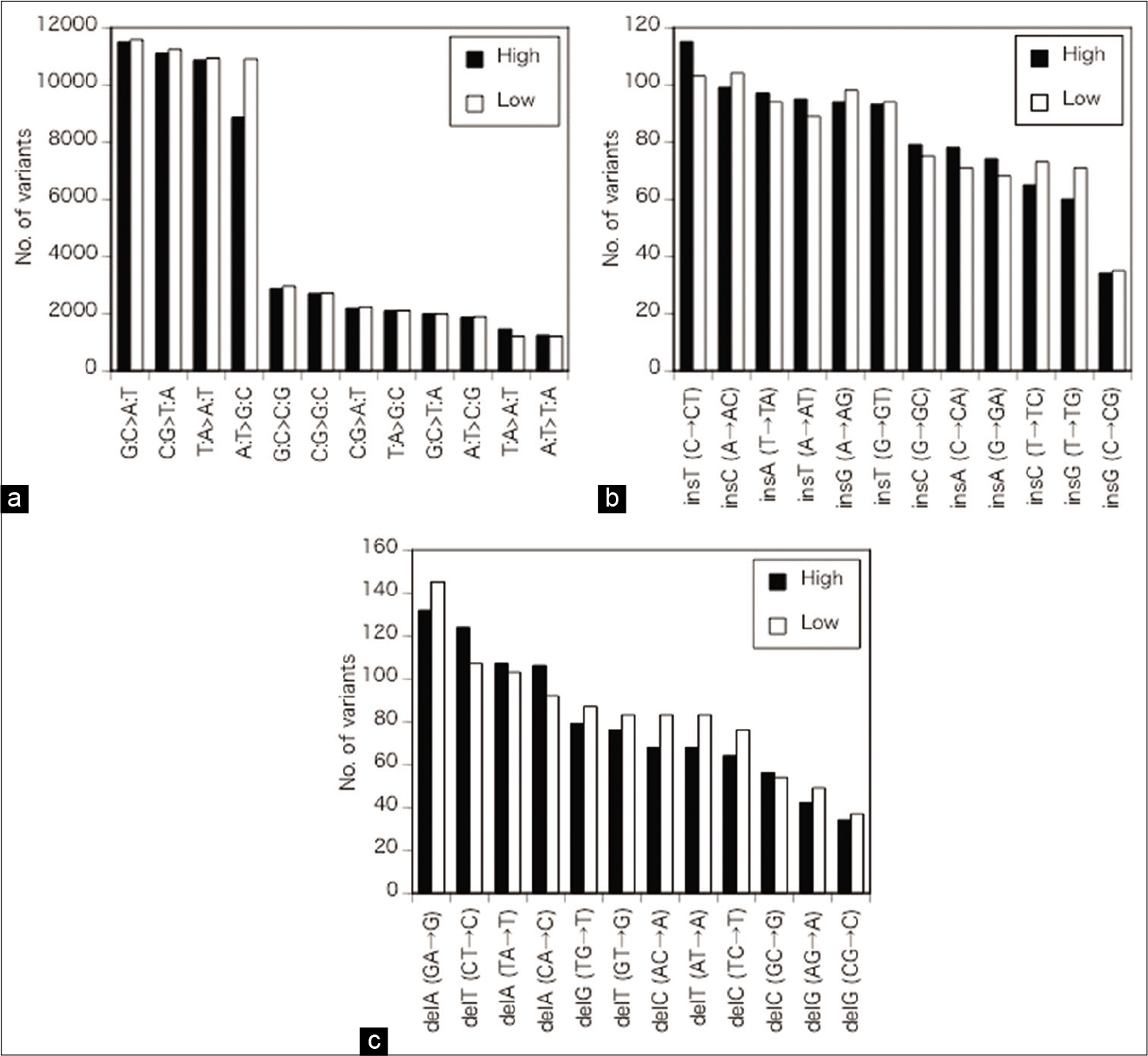
Figure 3:
(a) Details of the alterations of single nucleotides. Regarding SNVs, G:C>A:T and C:G>T:A transitions were the most frequent followed by T:A>A:T transversion and A:T>G:C transition. No significant differences in nucleotide changes between the high- and low- calcified plaque groups were apparent for the SNVs, except for a slight difference in A:T>G:C transversion. (b) Details of the alterations of single nucleotides. The most frequent insertions were insT after C and insC after A, followed by insA after T, insT after A, insG after A, and insT after G. No significant differences in nucleotide changes between the high- and low-calcified plaque groups were apparent for the ins. (c) Details of the alterations of single nucleotides. Among the deletions, delA after G and delT after C were the most common followed by delA after T and delA after C. No significant differences in nucleotide changes between the high- and low-calcified plaque groups were apparent for the deals. High: high-calcified plaques, low: low-calcified plaques.
Specific variants for the high- and low-calcified carotid plaques in the dbSNP
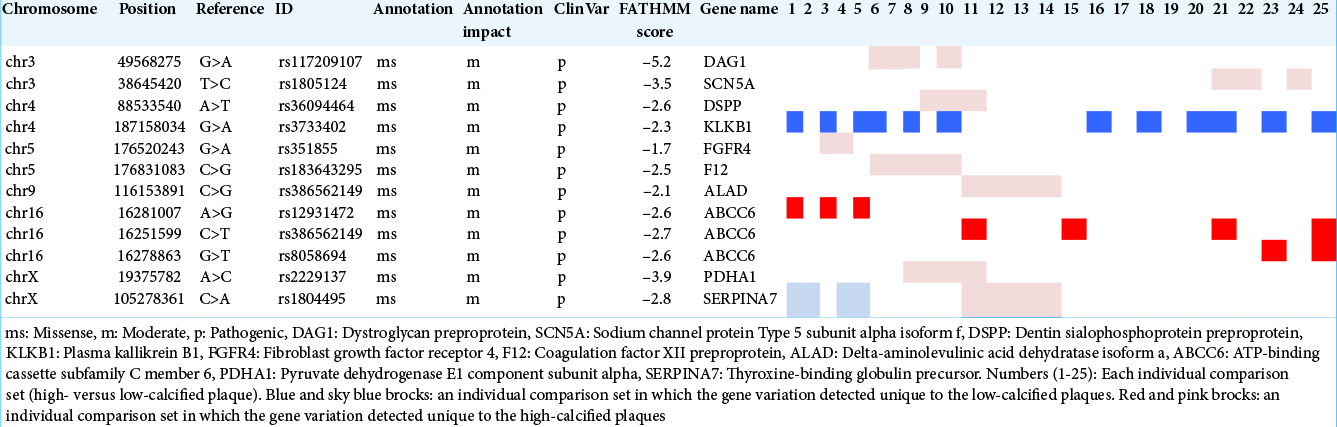
After the filtering was performed under the conditions described above, ten variations were extracted [
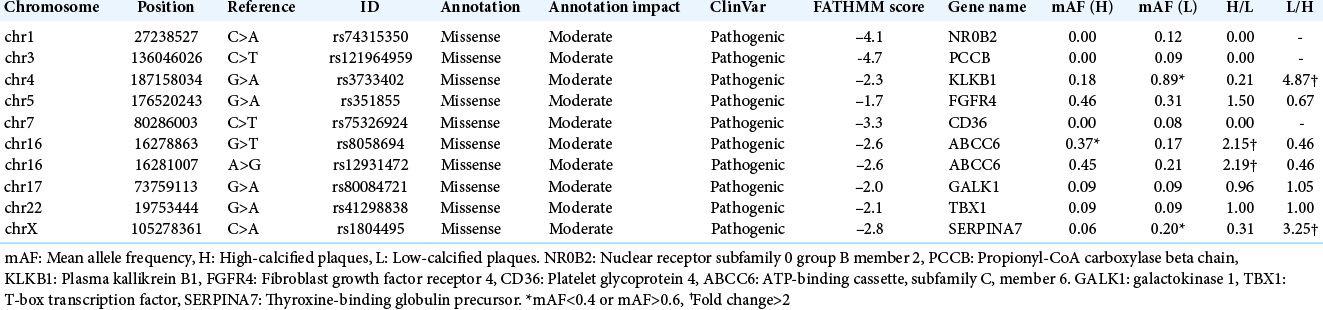
We then performed individual comparisons of high-calcified versus low-calcified specimens for 25 (5 × 5) sets. ABCC6 was chosen in 32% of the sets (8/25) for the high-calcified group. KLKB1 was selected in 48% of the sets (12/25) for the low-calcified group, but SERPINA7 was extracted only in 16% (4/25) [
Specific variants for high- and low-calcified carotid plaques outside of the dbSNP
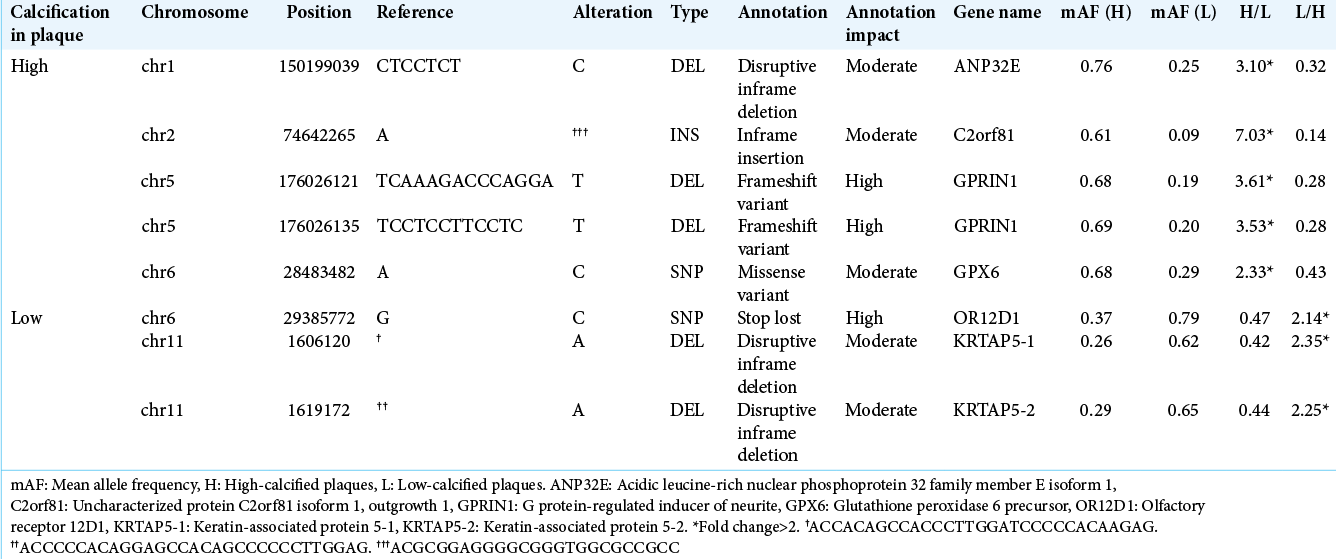
We attempted to identify novel variations by examining those that were not present in the dbSNP database. After the filtering under the conditions noted above was conducted, 98 variations in high-calcified plaques were those with a mean AF >0.6, and among them ANP32E, C2orfB1, GPRIN1, and GPX6 were variations of changes with a mean AF more than twofold that of the low-calcified group. On the other hand, 118 variations with a mean AF >0.6 were chosen in the low- calcified plaque group, and we selected OR12D1, KATAP5-1, and KATAP5-2 as variations of changes with a mean AF more than twofold that of the high-calcified group [
Our comparisons of individual samples between the high- and low-calcified plaque groups revealed five gene variations unique to high-calcified plaques and 13 variations unique to low-calcified plaques in >30% of all 25 sets [
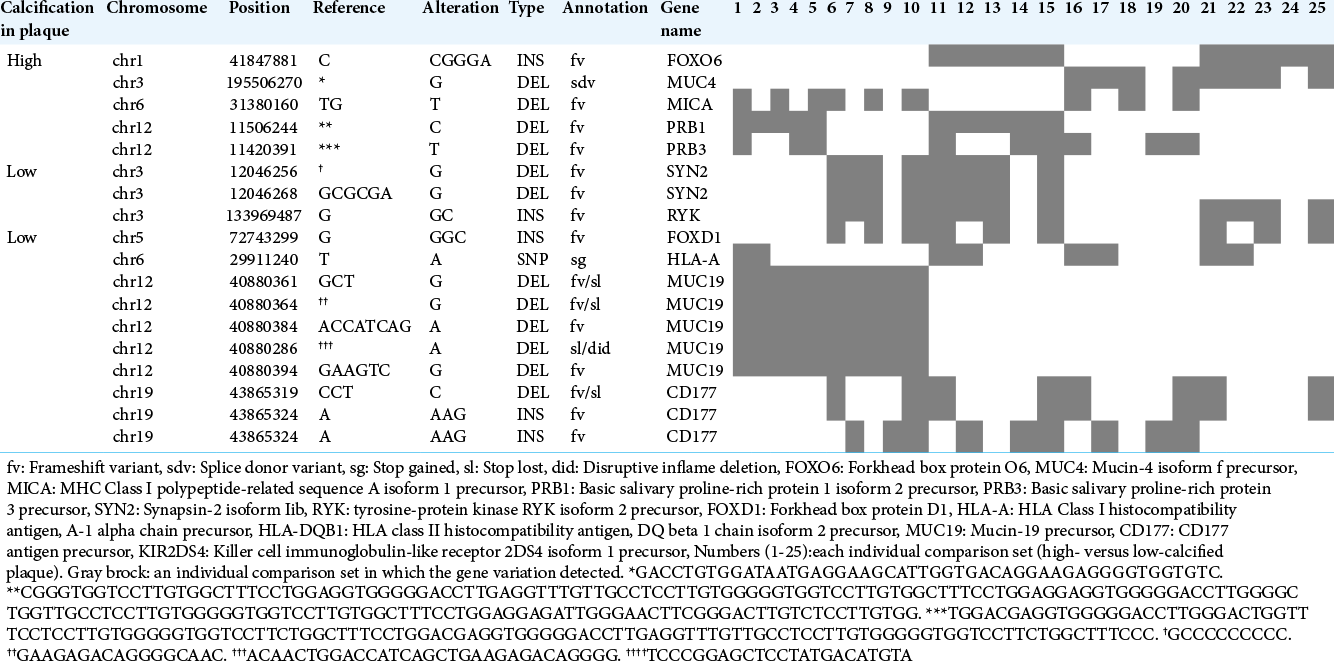
We conducted a trial for the functional enrichment of biological pathways for the above-noted detected genes using DAVID’s Functional Annotation Clustering. Regarding the seven genes detected by our group comparison, no enrichment was observed for the high- or low-calcified plaque group. [

DISCUSSION
The results of our present analyses demonstrated that in carotid plaques, G:C>A:T/C:G>T:A transitions as SNVs, insT after C/insC after A as insertions, and delA after G/delT after C as deletions were most frequently observed, but no significant difference was detected between the high- and low-calcified plaque groups in their proportion of base-pair substitution types. Compared to the other group, variants of ATP binding cassette subfamily C member 6 (ADCC6) were more commonly found in the high-calcified plaques and variants of KLKB1 were more commonly found in the low-calcified plaques. No new gene variants related to calcification or atherosclerosis was detected among those that were not registered in the dbSNP.
Types of base-pair substitutions in carotid plaques and the relation to calcification
To date, no specific and frequent substitutional types of SNPs or indels have been reported in the carotid artery, and little similar information is available for other aspects of atherosclerosis. In tumor studies, C:G>T:A transitions were the most frequent in parathyroid adenoma and non- functioning pituitary adenoma.[
Diversity is often observed among the cases in these tumor studies, whereas in the present study, we observed a relatively homogeneous distribution of base-pair substitution types among carotid plaque specimens. This might be attributable to the difference in heterogeneity between tumor and atherosclerosis specimens. It is conceivable that SNPs of G:C>A:T and C:G>T:A were the most frequent in our present investigation because it is generally easy for a transition in which purine/pyrimidine bases turn into the same bases to occur compared to a transversion that is a substitution between purine and pyrimidine bases. It is noteworthy that the proportions of the types of base-pair substitutions for SNPs as well as indels were mostly similar irrespective of the amount of calcification, except for the slight difference in A:T>G:C transition.
Previous genetic variant studies for atherosclerosis
Many genetic association investigations have attempted to determine the role(s) of certain gene mutations (especially SNPs) for atherosclerosis by performing GWAS or WES in large populations and meta-analyses, but the results conflict. Concerning GWASs for carotid atherosclerosis, the associated candidate genes were MMP-3 and other MMPs, CDKN2A/2B,IL-6/10, APOE, ACE, PON-1, and others.[
Most of these previous studies sought to identify hereditary gene variations such as SNPs derived from the germline by investigating samples from the peripheral blood of patients. Several SNPs identified by exhaustive searches and a GWAS or WES were used to confirm the association in other conditions, diseases, or cohorts. In the present study, we extracted the whole exome from affected carotid plaque specimens and compared the genome variations in high- and low-calcified carotid plaques based on calcium scores to detect the variations that were specific to each group. We excluded gene variations with 0.4≤ AF≤0.6, which may be highly likely to have a germline derivation from the ideal AF 0.5, to narrow the obtained data to somatic acquired information as much as possible.
The relationship between detected gene variations and calcification/atherosclerosis in carotid plaques
We extracted specific gene variations for each of the two calcium score-based classified plaque groups. Variations of ADCC6 gene, also known as multidrug resistance-associated protein 6, were more frequently detected in the present high-calcified plaque group; ADCC6 stimulates the release of adenosine triphosphate (ATP). Pyrophosphate broken down from ATP functions as an inhibitor of the production of hydroxyapatite crystals and regulates calcification and mineralization.[
been reported to be associated with generalized arterial calcification of infancy (GACI) which exhibits abnormal calcification of arteries.[
Plasma kallikrein encoded by KLKB1 – variations of which we observed more commonly in the low-calcified plaque group – cleaves high-molecular-weight kininogen to release bradykinin, which plays crucial roles in the vascular regulation of atherosclerosis by vasodilation and in the prevention of cell proliferation through the kinin-kallikrein system.[
The search for calcified-plaque-specific variants not listed in the dbSNP
The above gene variants were carefully selected using previously obtained information about variants that are likely to be involved in amino acid/protein alterations and to be pathogenic. Herein we attempted to find novel variations that are specific to high- or low-calcified plaques that are not yet in the dbSNP database. Seven mutated genes from our group comparison and 12 genes from the individual comparisons were identified and applied to the functional analysis. Unfortunately, no enrichment for gene variants from the group comparison and no genes with a high- enough enrichment score from the individual comparisons were detected, presumably mainly due to the small sample size. Larger cohort studies might be helpful to address this.
The clinical significance of investigations of calcified carotid plaques
In general, the culprit plaques have been intently investigated because the results may be directly useful in the future for symptom alleviation and preventive treatments for symptomatic plaques. The prior investigations included studies of gene regulation therapies that inhibit promoters of the proliferation of vascular smooth muscle cells or angiogenesis using miRNAs.[
As demonstrated in the previous studies[
Limitations
The main limitation of the study is the small sample number. In addition, we cannot clearly declare whether the gene variations extracted herein were somatic or germline- derived. We harvested the genome from plaques with the acquired atheromatous change, not from peripheral blood, as in the prior SNP studies for rare variants. We may have thus more opportunities to obtain somatic-prone genetic variations. We also excluded the variants with 0.4 ≤AF ≤0.6 that were likely to have a germline origin in order to narrow down the genetic information to somatic information as much as possible, avoiding extra invasive interventions to the patients. More precise information would have been obtained, however, by collecting additional peripheral blood samples or other normal tissues and comparing them with the carotid plaque results. We are designing a further investigation in consideration of these points.
CONCLUSION
Our findings demonstrated the precise profile of base-pair substitutions in carotid plaques. No significant differences were detected between the high- and low-calcified plaques. However, genetic variants in ADCC6 relating to vascular calcification for high-calcified plaques, and in KLKB1 encoding kallikrein associated with vascular regulation of atherosclerosis for low-calcified plaques were more frequently discovered compared to the other group. These results may contribute to a better understanding of the genetic signatures of calcium modulation and cellular activity in carotid plaques.
Declaration of patient consent
Institutional Review Board permission obtained for the study.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Bezemer ID, Bare LA, Doggen CJ, Arellano AR, Tong C, Rowland CM. Gene variants associated with deep vein thrombosis. JAMA. 2008. 299: 1306-14
2. Chen LJ, Lim SH, Yeh YT, Lien SC, Chiu JJ. Roles of microRNAs in atherosclerosis and restenosis. J Biomed Sci. 2012. 19: 79
3. Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012. 6: 80-92
4. De Vries PS, Yu B, Feofanova EV, Metcalf GA, Brown MR, Zeighami AL. Whole-genome sequencing study of serum peptide levels: The atherosclerosis risk in communities study. Hum Mol Genet. 2017. 26: 3442-50
5. Ewence AE, Bootman M, Roderick HL, Skepper JN, McCarthy G, Epple M. Calcium phosphate crystals induce cell death in human vascular smooth muscle cells: A potential mechanism in atherosclerotic plaque destabilization. Circ Res. 2008. 103: e28-34
6. Fang Y, Ma X, Zeng J, Jin Y, Hu Y, Wang J. The profile of genetic mutations in papillary thyroid cancer detected by whole exome sequencing. Cell Physiol Biochem. 2018. 50: 169-78
7. Forgo B, Medda E, Hernyes A, Szalontai L, Tarnoki DL, Tarnoki AD. Carotid artery atherosclerosis: A review on heritability and genetics. Twin Res Hum Genet. 2018. 21: 333-46
8. Franceschini N, Giambartolomei C, De Vries PS, Finan C, Bis JC, Huntley RP. GWAS and colocalization analyses implicate carotid intima-media thickness and carotid plaque loci in cardiovascular outcomes. Nat Commun. 2018. 9: 5141
9. Germain DP. Pseudoxanthoma elasticum. Orphanet J Rare Dis. 2017. 12: 85
10. Heit JA, Cunningham JM, Petterson TM, Armasu SM, Rider DN, De Andrade M. Genetic variation within the anticoagulant, procoagulant, fibrinolytic and innate immunity pathways as risk factors for venous thromboembolism. J Thromb Haemost. 2011. 9: 1133-42
11. Katano H, Mase M, Nishikawa Y, Yamada K. Calcified carotid plaques show double symptomatic peaks according to Agatston calcium score. J Stroke Cerebrovasc Dis. 2015. 24: 1341-50
12. Katano H, Mase M, Nishikawa Y, Yamada K. Surgical treatment for carotid stenoses with highly calcified plaques. J Stroke Cerebrovasc Dis. 2014. 23: 148-54
13. Katano H, Yamada K. Analysis of calcium in carotid plaques with Agatston scores for appropriate selection of surgical intervention. Stroke. 2007. 38: 3040-4
14. Katsuda I, Maruyama F, Ezaki K, Sawamura T, Ichihara Y. A new type of plasma prekallikrein deficiency associated with homozygosity for Gly104Arg and Asn124Ser in apple domain 2 of the heavy-chain region. Eur J Haematol. 2007. 79: 59-68
15. Kwee RM. Systematic review on the association between calcification in carotid plaques and clinical ischemic symptoms. J Vasc Surg. 2010. 51: 1015-25
16. Le Saux O, Martin L, Aherrahrou Z, Leftheriotis G, Váradi A, Brampton CN. The molecular and physiological roles of ABCC6: More than meets the eye. Front Genet. 2012. 3: 289
17. Li HW, Shen M, Gao PY, Li ZR, Cao JL, Zhang WL. Association between ADAMTS7 polymorphism and carotid artery plaque vulnerability. Medicine (Baltimore). 2019. 98: e17438
18. Li Q, Brodsky JL, Conlin LK, Pawel B, Glatz AC, Gafni RI. Mutations in the ABCC6 gene as a cause of generalized arterial calcification of infancy: Genotypic overlap with pseudoxanthoma elasticum. J Invest Dermatol. 2014. 134: 658-65
19. Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018. 50: 524-37
20. Moitra K, Garcia S, Jaldin M, Etoundi C, Cooper D, Roland A. ABCC6 and pseudoxanthoma elasticum: The face of a rare disease from genetics to advocacy. Int J Mol Sci. 2017. 18: 1488
21. Nandalur KR, Baskurt E, Hagspiel KD, Finch M, Phillips CD, Bollampally SR. Carotid artery calcification on CT may independently predict stroke risk. AJR Am J Roentgenol. 2006. 186: 547-52
22. Newey PJ, Nesbit MA, Rimmer AJ, Attar M, Head RT, Christie PT. Whole-exome sequencing studies of nonhereditary (sporadic) parathyroid adenomas. J Clin Endocrinol Metab. 2012. 97: E1995-2005
23. Newey PJ, Nesbit MA, Rimmer AJ, Head RA, Gorvin CM, Attar M. Whole-exome sequencing studies of nonfunctioning pituitary adenomas. J Clin Endocrinol Metab. 2013. 98: E796-800
24. Nitschke Y, Baujat G, Botschen U, Wittkampf T, Du Moulin M, Stella J. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am J Hum Genet. 2012. 90: 25-39
25. Pan W, Zhou L, Ge M, Zhang B, Yang X, Xiong X. Whole exome sequencing identifies lncRNA GAS8-AS1 and LPAR4 as novel papillary thyroid carcinoma driver alternations. Hum Mol Genet. 2016. 25: 1875-84
26. Parreira B, Cardoso JC, Costa R, Couto AR, Bruges-Armas J, Power DM. Persistence of the ABCC6 genes and the emergence of the bony skeleton in vertebrates. Sci Rep. 2018. 8: 6027
27. Petretta M, Daniele S, Acampa W, Imbriaco M, Pellegrino T, Messalli G. Prognostic value of coronary artery calcium score and coronary CT angiography in patients with intermediate risk of coronary artery disease. Int J Cardiovasc Imaging. 2012. 28: 1547-56
28. Polonsky TS, McClelland RL, Jorgensen NW, Bild DE, Burke GL, Guerci AD. Coronary artery calcium score and risk classification for coronary heart disease prediction. JAMA. 2010. 303: 1610-6
29. Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification in vitro Evidence for initiation of vascular calcification by apoptotic bodies. Circ Res. 2000. 87: 1055-62
30. Rutsch F, Nitschke Y, Terkeltaub R. Genetics in arterial calcification: Pieces of a puzzle and cogs in a wheel. Circ Res. 2011. 109: 578-92
31. Schunkert H, Von Scheidt M, Kessler T, Stiller B, Zeng L, Vilne B. Genetics of coronary artery disease in the light of genome-wide association studies. Clin Res Cardiol. 2018. 107: 2-9
32. Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013. 34: 57-65
33. Tibaut M, Caprnda M, Kubatka P, Sinkovič A, Valentova V, Filipova S. Markers of atherosclerosis: Part 2-genetic and imaging markers. Heart Lung Circ. 2019. 28: 678-89
34. Vojinovic D, Kavousi M, Ghanbari M, Brouwer RW, Van Rooij JG, Van Den Hout MC. Whole-genome linkage scan combined with exome sequencing identifies novel candidate genes for carotid intima-media thickness. Front Genet. 2018. 9: 420
35. Yu B, Zheng Y, Alexander D, Morrison AC, Coresh J, Boerwinkle E. Genetic determinants influencing human serum metabolome among African Americans. PLoS Genet. 2014. 10: e1004212
36. Zhang Y, Wang L, Zhang Z, Zhang Z, Zhou S, Cao L. Shared and discrepant susceptibility for carotid artery and aortic arch calcification: A genetic association study. Atherosclerosis. 2015. 241: 371-5

