

- Department of Neurosurgery, Hamad General Hospital, Doha, Qatar.
- Department of Histopathology, Hamad General Hospital, Doha, Qatar.
- Department of Neuroradiology, Hamad General Hospital, Doha, Qatar.
Correspondence Address:
Saleh Salah Safi
Department of Histopathology, Hamad General Hospital, Doha, Qatar.
DOI:10.25259/SNI_391_2020
Copyright: © 2020 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Saleh Salah Safi1, Khaled Murshed2, Arshad Ali1, Surjith Vattoth3, Abdulrazzaq Haider2, Issam Al-Bozom2. Rosai-Dorfman disease of cranial and spinal origin - A case series. 18-Sep-2020;11:298
How to cite this URL: Saleh Salah Safi1, Khaled Murshed2, Arshad Ali1, Surjith Vattoth3, Abdulrazzaq Haider2, Issam Al-Bozom2. Rosai-Dorfman disease of cranial and spinal origin - A case series. 18-Sep-2020;11:298. Available from: https://surgicalneurologyint.com/surgicalint-articles/10264/
Date of Submission
29-Jun-2020
Date of Acceptance
28-Aug-2020
Date of Web Publication
18-Sep-2020
Abstract
Background: Rosai-Dorfman disease (RDD) is an idiopathic nonneoplastic lymphadenopathy disorder which is characterized by lymph node enlargement, but it may also presents primarily involving a variety of extranodal sites, including central nerves system and craniospinal axis. This study reports five cases of craniospinal RDD, with review of epidemiology, clinical presentation, imaging, and histopathological features with current management strategies.
Case Description: Five cases of RDD are diagnosed at Hamad General Hospital, Qatar, during 2013–2018. Two cases had dural-based cranial lesions with overlying cranial involvement while three cases were having extradural thoracic spine lesions. All cases underwent surgical intervention and confirmed by histopathology.
Conclusion: Craniospinal RDD is a rare clinical presentation and poses significant diagnostic challenges preoperatively due to its similarity with other neoplastic or inflammatory diseases. Surgical option to remove compressive neural pathology provides a good clinical outcome with no recurrence in long-term follow-up.
Keywords: Central nerves system, Disease, Dural based, Lymphadenopathy, Meningioma, Resection. RosaiDorfman, Sinus histiocytosis, Spinal
INTRODUCTION
Rosai-Dorfman disease (RDD), or sinus histiocytosis with massive lymphadenopathy, is a benign, idiopathic histiocytic proliferative disease with pathognomonic histological and immunohistochemical characteristics.[
Extranodal lesions have been reported at various sites including the central nerves system (CNS) and the bone; however, bone involvement was reported to be <10% with only two cases reported involving the skull bone to date.[
CASES DESCRIPTION
Case No.1
A 28-year-old female patient presented with 10 days history of increasing headache, associated with weakness of the right upper and lower limbs. On clinical examination, she had normal vital signs, fully conscious and oriented, right hemiparesis Grade 4 (Medical Research Council Scale). There were no signs of lymphadenopathy. Computerized tomography (CT) scan and magnetic resonance imaging (MRI) of the brain revealed a calvarial epicentered mass lesion with sharp beveled-edged bony defect, extracranial and intracranial extradural and sulcal extension, and a surrounding brain edema. The lesion displayed iso- to low signal intensity on T2W, intermediate signal on T1W MRI, postcontrast enhancement, reduced relative cerebral blood volume (rCBV) on perfusion MRI, and no diffusion restriction [
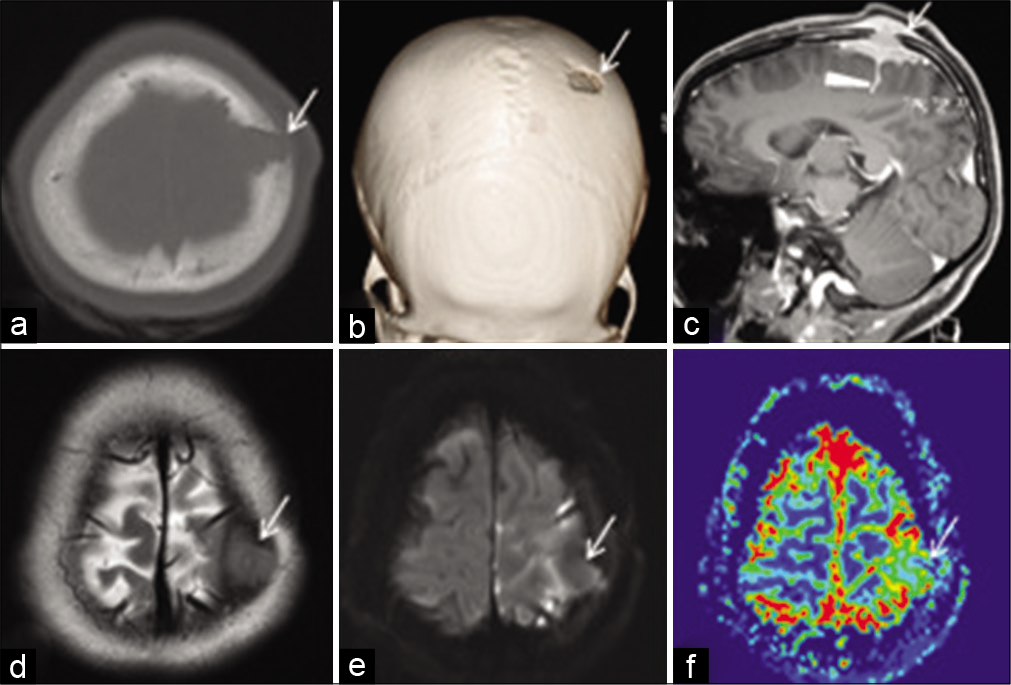
Figure 1:
Axial (a) and 3D volume rendered (b) computerized tomography scan bone window images show a sharp defect in the anterior left parietal bone with beveled edges (arrow). Sagittal postcontrast 3D T1 MPRAGE magnetic resonance imaging (MRI) (c) shows enhancing soft tissue in the scalp and intracranial extra-axial location extending across and filling the calvarial defect (arrow); with extension of enhancing soft tissue into the underlying sulcus (arrowhead). Axial T2W MRI (d) shows that the lesion (arrow) is iso to slightly hypointense to cortex. Axial DWI MRI (e) shows no diffusion restriction within the lesion (arrow). Axial perfusion MRI relative cerebral blood volume (rCBV) image (f) shows reduced rCBV within the lesion (arrow) compared with the adjacent cortex.
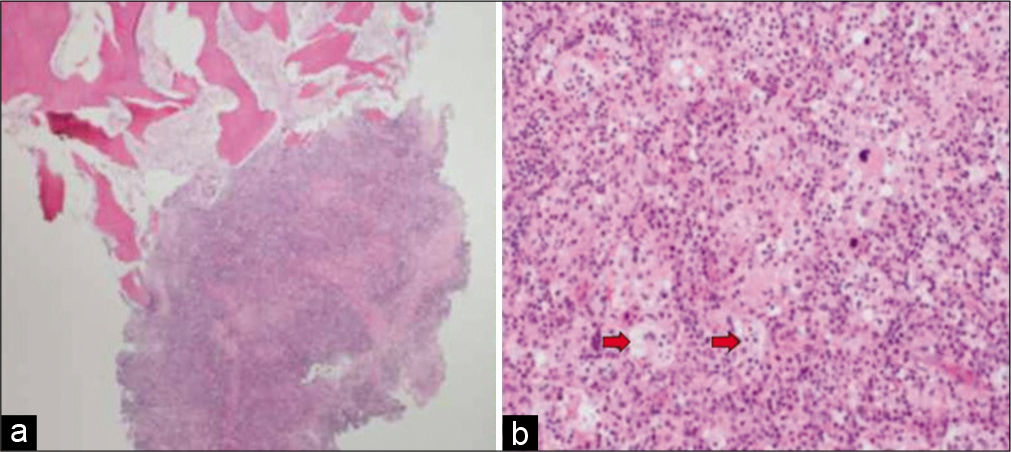
Figure 2:
(a) Photomicrograph depicting bony trabeculae infiltrated by Rosai-Dorfman disease (hematoxylin and eosin stain ×40. (b) High-power view shows histiocytic proliferation along with diffuse lymphoplasmacytic infiltrate. Some histiocytes are large with emperipolesis (red arrows) (hematoxylin and eosin stain ×200).
Case No.2
A 25-year-old male patient presented to emergency department (ED) after found unconscious, with an unwitnessed seizure activity. On arrival in ED, he regained his consciousness but had retrograde amnesia. On examination, he had normal vital signs and his Glasgow Coma Scale (GCS) was 15/15 with no neurological deficits. CT scan showed left posterior frontal hypodense lesion. MRI revealed a left inferior parietal extra-axial mass lesion displaying predominant low signal intensity on T2W, intermediate signal on T1W MRI images, postcontrast enhancement, reduced rCBV on perfusion MRI, and no diffusion restriction. There were contiguous enhancing dural tails and a T2 hypointense enhancing tiny nodular focus in the adjacent brain. There was perilesional brain edema. No bony involvement was seen [
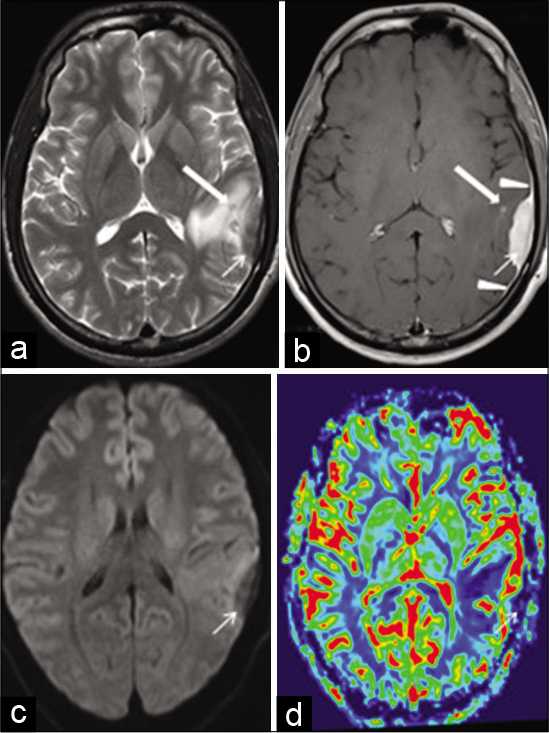
Figure 3:
Axial T2W magnetic resonance imaging (MRI) (a) shows a left inferior parietal extra-axial soft-tissue lesion (arrow) with surrounding hyperintense edema. The lesion is hypointense to cortex and there is also a tiny hypointense nodule in the adjacent brain (thick arrow) with intact calvarium. Axial postcontrast TW MRI (b) shows uniform enhancement of the lesion (arrow). Note the contiguous enhancing dural tails (arrowheads) and enhancement of the tiny nodular focus in the adjacent brain (thick arrow). Axial DWI MRI (c) shows no diffusion restriction within the lesion (arrow). Axial perfusion MRI relative cerebral blood volume (rCBV) image (d) shows reduced rCBV within the lesion (arrow) compared with the adjacent brain.
Case No.3
A 25-year-old male patient presented with complaints of progressive lower limbs weakness and difficulty in walking for the past 10 days. On examination, his GCS was 15/15 and he had bilateral lower limb weakness Grade 3/5, hyperreflexia, and muscle spasticity with sensory level of thoracic (T) vertebra no. 6. MRI of spine showed enhancing right paravertebral soft-tissue lesion extending from cervical (C) vertebra No. 7 through T4 vertebral levels with extradural extension encasing the spinal cord with cord edema and patchy areas of myelitis. There was no significant vertebral bony involvement [
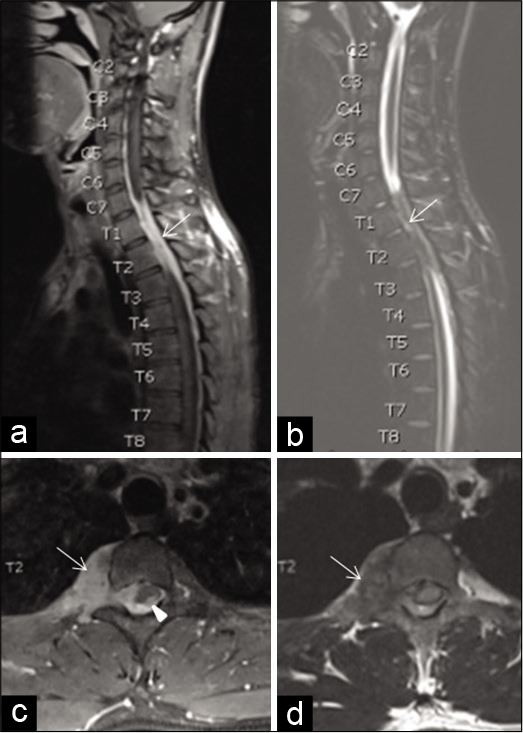
Figure 5:
Sagittal postcontrast T1W magnetic resonance imaging (MRI) of the cervicothoracic spine (a) shows a right-sided enhancing epidural soft-tissue lesion (arrow) which appears hypointense on T2W-STIR MRI (b) at C7-T4 spinal canal levels. Axial postcontrast T1W MRI (c) shows extension through the right neural foramen with a sizeable paravertebral soft-tissue component (arrow). Note a tiny enhancing component within the spinal cord parenchyma (arrowhead). Axial T2WI (d) shows hypointensity of the paraspinal lesion and diffuse spinal cord hyperintensity. No vertebral bony involvement is seen.
Case No.4
A 52-year-old male patient presented progressive difficulty in walking and urinary retention for the past 3 days. On examination, he had bilateral lower limb weakness Grade 2/5, with bilateral Babinski’ sign extensor bilaterally and muscle spasticity with sensory level of T6 and lax anal tone. MRI of the spine showed a large epidural enhancing soft-tissue lesion nearly filling the whole spinal canal at T4 level with dural tails superiorly and inferiorly. The lesion produced significant compression and focal displacement of the thoracic spinal cord to the left anterolateral aspect. In addition, there was a slightly T1 and T2 hyperintense enhancing lesion within the T4 vertebral body, which could bring in the differential diagnosis of vertebral bony and spinal canal metastatic deposits. However, a correlative CT scan image clearly showed that the bony lesion was an incidental vertebral body hemangioma [
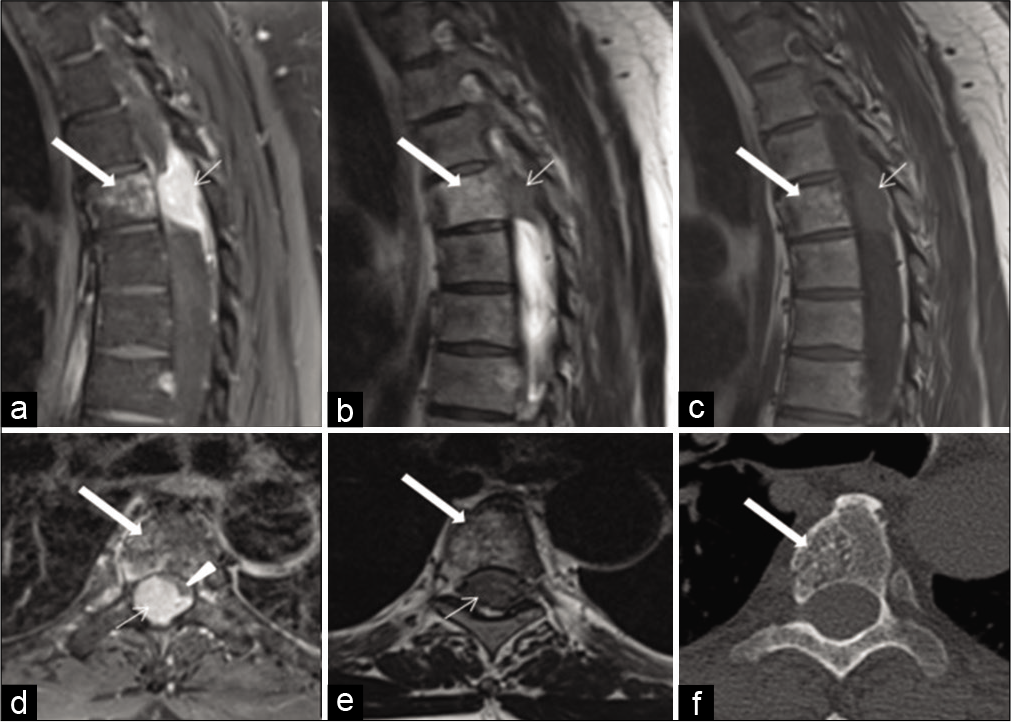
Figure 7:
Sagittal postcontrast T1W magnetic resonance imaging (MRI) of the thoracic spine (a) shows an enhancing epidural soft-tissue lesion nearly filling the spinal canal (arrow) with dural tails superiorly and inferiorly, which appears hypointense on T2W (b) and T1W (c) MRI at T4 vertebral level. Axial postcontrast MRI (d) shows severe compression and displacement of the spinal cord to the left anterolateral aspect (arrowhead) by the enhancing lesion (arrow). Axial T2W MRI (e) shows hypointensity of the lesion filling the canal (arrow) and no hyperintense fluid signal of surrounding CSF is seen as it is obliterated. All of the above MR sequences show a slightly T1 and T2 hyperintense, enhancing lesion within the T4 vertebral body (thick arrow), which could introduce the differential diagnosis of vertebral bony and spinal canal metastatic components. Axial bone window computerized tomography scan (f) image, however, clearly shows that the bony lesion is just an incidental vertebral body hemangioma (thick arrow) with characteristic “polka-dot” appearance of its vertical striations.
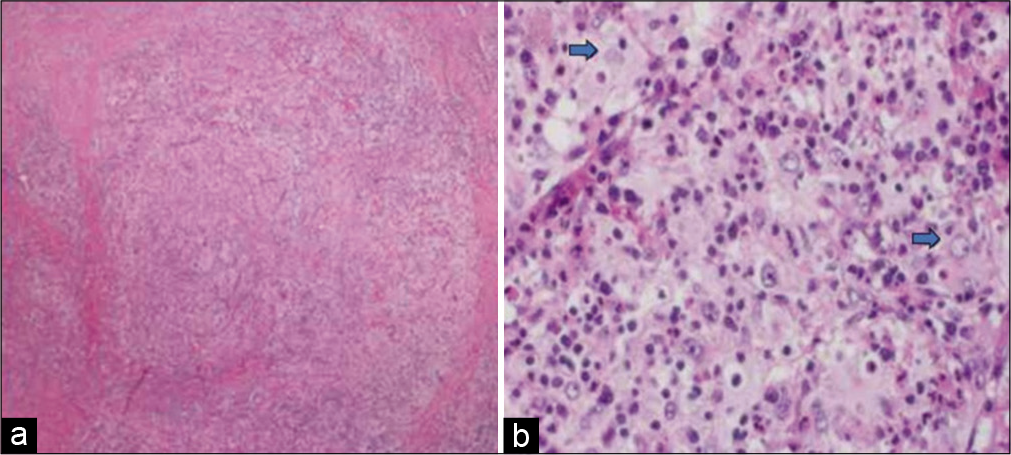
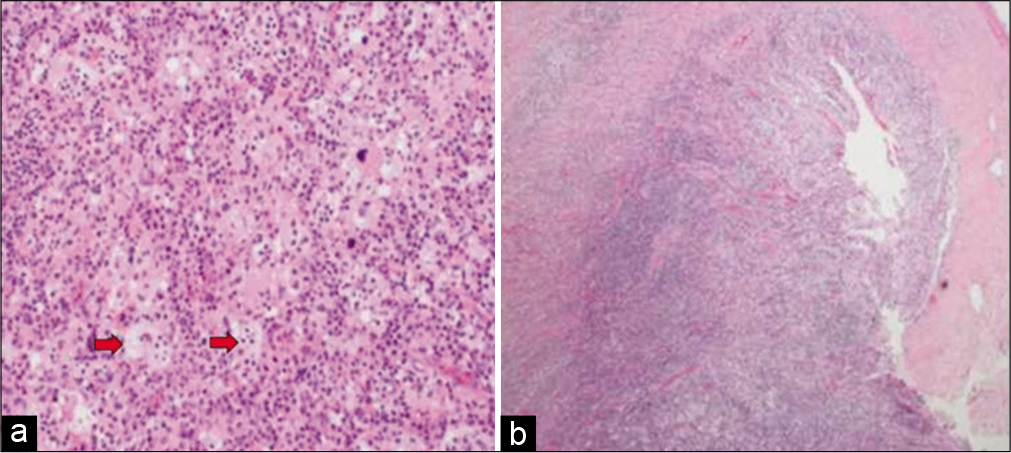
Figure 8:
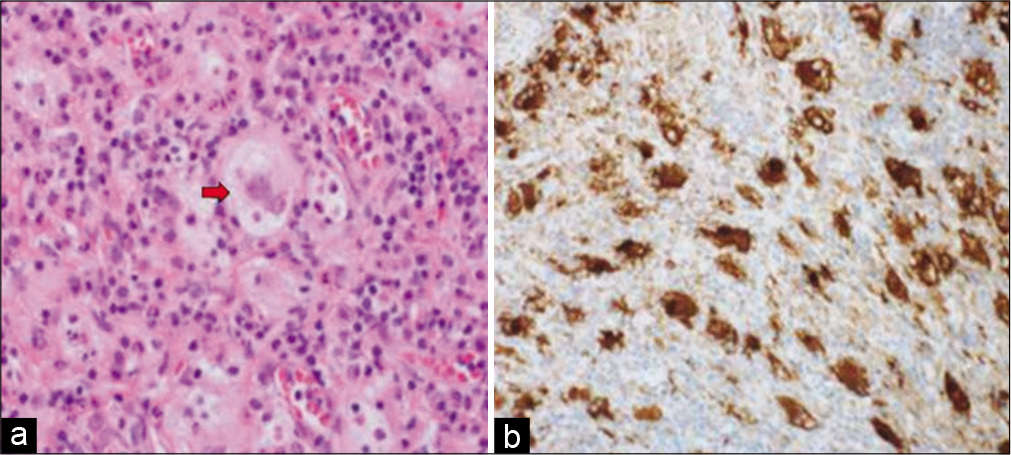
(a) Photomicrograph showing diffuse lymphohistiocytic proliferation in collagenous stroma (hematoxylin and eosin stain ×40). (b) High-power view shows histiocytic proliferation along with diffuse lymphoplasmacytic infiltrate. Some histiocytes with engulfed intact lymphocytes are present (blue arrows) (hematoxylin and eosin stain ×200).
Case No.5
A 40-year-old Qatari male presented with 1-day history of bilateral lower limbs weakness associated with urinary incontinence. On examination, he had bilateral lower limb weakness, more pronounced distally along with bilateral spasticity and pinprick sensory level at T8. MRI of the spine showed a large epidural enhancing soft-tissue lesion nearly filling the whole spinal canal extending from T6 to T11, with dural tails superiorly and inferiorly. The lesion produced significant compression and focal displacement of the thoracic spinal cord to the left anterolateral aspect. The patient underwent T7-11 laminotomy and excision of extradural mass. Histopathology showed a dura-based lesion composed of histiocytic proliferation with lymphoplasmacytic infiltrate along with lymphophagocytosis, compatible with RDD. Postoperative MRI showed no residual tumoral tissue. The patient showed improvement in his power postoperatively, subjected to with rehabilitation and was discharged with mild spasticity of both lower limbs with power 5/5 on the latest outpatient follow-up visit.
DISCUSSION
RDD is a benign, nonneoplastic histiocytic proliferative disease characterized by prominent cervical lymph node enlargement; however, cases also illustrated that RDD might involve multiple organ systems and be indeed fatal.[
The etiology of RDD remains unclear since its recognition in 1969 and infectious etiology is the aspect that attracts most of the attention.[
The patient with cranial RDD is often free from the systemic symptoms and may present with only neurological symptoms.[
Intracranial or spinal RDD usually presents as extra-axial dural based lesion which is iso- to hypointense to gray matter on T1 and T2 W MRI, usually solitary and rarely multiple.[
Histologically, RDD is characterized by proliferation of histiocytes that are large with round nuclei and abundant pale cytoplasm, along with diffuse lymphoplasmacytic infiltrate.[
Spontaneous resolution and stable asymptomatic disease are observed in about of 90% of cases. In cases without CSN involvement, surgical resection is justified both to remove compressive lesion and establish a histopathological diagnosis.[
Long-term prognosis is believed to be associated with a number of nodal groups and extranodal systems involved, indicating that patients with extranodal lesions will probably have a poorer prognosis than those with nodal diseases.[
CONCLUSION
Craniospinal RDD is a rare clinical condition and its clinical presentation mimics other compressive pathology. In isolated craniospinal involvement, it poses a significant diagnostic challenge due to its similarity with other neoplastic and inflammatory pathologies involving craniospinal axis. Some peculiar neuroradiological features may help to suspect a preoperative diagnosis; however, histopathological distinction remains the standard of diagnosis. Surgical option is deemed necessary to treat compressive parenchymal clinical presentation and establish a definitive diagnosis. Craniospinal RDD has shown a good clinical outcome with complete neuroradiological resolution and no recurrence in long-term follow-up.
Declaration of patient consent
Institutional Review Board permission obtained for the study.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Abdelwahab IF, Klein MJ, Springfield DS, Hermann G. A solitary lesion of talus with mixed sclerotic and lytic changes: Rosai-Dorfman disease of 25 years’ duration. Skeletal Radiol. 2004. 33: 230-3
2. Adeleye AO. Diagnosis and management of Rosai-Dorfman disease involving the central nervous system. Neurol Res. 2010. 32: 572-8
3. Allegranza A, Barbareschi M, Solero CL, Fornari M, Lasio G. Primary lymphohistiocytic tumour of bone: A primary osseous localization of Rosai-Dorfman disease. Histopathology. 1991. 18: 83-6
4. Alqanatish JT, Houghton K, Bond M, Senger C, Tucker LB. Rituximab treatment in a child with Rosai-Dorfman disease and systemic lupus erythematosus. J Rheumatol. 2010. 37: 1783-4
5. Al-Saad K, Thorner P, Ngan BY, Gerstle JT, Kulkarni AV, Babyn P. Extranodal Rosai-Dorfman disease with multifocal bone and epidural involvement causing recurrent spinal cord compression. Pediatr Dev Pathol. 2005. 8: 593-8
6. Andriko JA, Morrison A, Colegial CH, Davis BJ, Jones RV. Rosai-Dorfman disease isolated to the central nervous system: A report of 11 cases. Mod Pathol. 2001. 14: 172-8
7. Becroft DM, Dix MR, Gillman JC, MacGregor BJ, Shaw RL. Benign sinus histiocytosis with massive lymphadenopathy: Transient immunological defects in a child with mediastinal involvement. J Clin Pathol. 1973. 26: 463-9
8. Buchino JJ, Byrd RP, Kmetz DR. Disseminated sinus histiocytosis with massive lymphadenopathy: Its pathologic aspects. Arch Pathol Lab Med. 1982. 106: 13-6
9. Destombes P. Adenitis with lipid excess, in children or young adults, seen in the Antilles and in Mali (4 cases). Bull Soc Pathol Exot Filiales. 1965. 58: 1169-75
10. Fatobene G, Haroche J, Hélias-Rodzwicz Z, Charlotte F, Taly V, Ferreira AM. BRAF V600E mutation detected in a case of Rosai-Dorfman disease. Haematologica. 2018. 103: e377-9
11. Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): Review of the entity. Semin Diagn Pathol. 1990. 7: 19-73
12. Geara AR, Ayoubi MA, Achram MC, Chamseddine NM. Rosai-Dorfman disease mimicking neurofibromatosis: Case presentation and review of the literature. Clin Radiol. 2004. 59: 625-30
13. Gupta K, Bagdi N, Sunitha P, Ghosal N. Isolated intracranial Rosai-Dorfman disease mimicking meningioma in a child: A case report and review of the literature. Br J Radiol. 2011. 84: e138-41
14. Hadjipanayis CG, Bejjani G, Wiley C, Hasegawa T, Maddock M, Kondziolka D. Intracranial Rosai-Dorfman disease treated with microsurgical resection and stereotactic radiosurgery. Case report. J Neurosurg. 2003. 98: 165-8
15. Hargett C, Bassett T. Atypical presentation of sinus histiocytosis with massive lymphadenopathy as an epidural spinal cord tumor: A case presentation and literature review. J Spinal Disord Tech. 2005. 18: 193-6
16. Hingwala D, Neelima R, Kesavadas C, Thomas B, Kapilamoorthy TR, Radhakrishnan VV. Advanced MRI in Rosai-Dorfman disease: Correlation with histopathology. J Neuroradiol. 2011. 38: 113-7
17. Idir I, Cuvinciuc V, Uro-Coste E, Penna M, Boetto S, Cognard C. MR perfusion of intracranial Rosai-Dorfman disease mimicking meningioma. J Neuroradiol. 2011. 38: 133-4
18. Kattner KA, Stroink AR, Roth TC, Lee JM. Rosai-Dorfman disease mimicking parasagittal meningioma: Case presentation and review of literature. Surg Neurol. 2000. 53: 452-7
19. Lu LX, Della-Torre E, Stone JH, Clark SW. IgG4-related hypertrophic pachymeningitis: Clinical features, diagnostic criteria, and treatment. JAMA Neurol. 2014. 71: 785-93
20. Ludemann W, Banan R, Samii A, Koutzoglou M, Di Rocco C. Cerebral Rosai-Dorfman disease. Child Nerv Syst. 2015. 31: 529-32
21. Lungren MP, Petrella JR, Cummings TJ, Grant GA. Isolated intracranial Rosai-Dorfman disease in a child. AJNR Am J Neuroradiol. 2009. 30: E148-9
22. Raslan O, Ketonen LM, Fuller GN, Schellingerhout D. Intracranial Rosai-Dorfman disease with relapsing spinal lesions. J Clin Oncol. 2008. 26: 3087-9
23. Raveenthiran V, Dhanalakshmi M, Hayavadana Rao PV, Viswanathan P. Rosai-Dorfman disease: Report of a 3-year-old girl with critical review of treatment options. Eur J Pediatr Surg. 2003. 13: 350-4
24. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol. 1969. 87: 63-70
25. Sandoval-Sus JD, Sandoval-Leon AC, Chapman JR, Velazquez-Vega J, Borja MJ, Rosenberg S. Rosai-Dorfman disease of the central nervous system: Report of 6 cases and review of the literature. Medicine (Baltimore). 2014. 93: 165-75
26. Sita G, Guffanti A, Colombi M, Ferrari A, Neri A, Baldini L. Rosai-Dorfman syndrome with extranodal localizations and response to glucocorticoids: A case report. Haematologica. 1996. 81: 165-7
27. Utikal J, Ugurel S, Kurzen H, Erben P, Reiter A, Hochhaus A. Imatinib as a treatment option for systemic nonLangerhans cell histiocytoses. Arch Dermatol. 2007. 143: 736-40
28. Xu H, Zhang F, Lu F, Jiang J. Spinal Rosai-Dorfman disease: Case report and literature review. Eur Spine J. 2017. 26: 117-27
29. Yang X, Liu J, Ren Y, Richard SA, Zhang Y. Isolated intracranial Rosai-Dorfman disease mimicking petroclival meningioma in a child. Case report and review of the literature. Medicine (Baltimore). 2017. 96: e8754
30. Zhu H, Qiu LH, Dou YF, Wu JS, Zhong P, Jiang CC. Imaging characteristics of Rosai-Dorfman disease in the central nervous system. Eur J Radiol. 2012. 81: 1265-72

