

- Division of Neurological Surgery, University of Missouri School of Medicine, Columbia, Missouri, USA
- Department of Orthopedic Surgery, University of Missouri School of Medicine, Columbia, Missouri, USA
Correspondence Address:
Tomoko Tanaka
Division of Neurological Surgery, University of Missouri School of Medicine, Columbia, Missouri, USA
DOI:10.4103/sni.sni_463_16
Copyright: © 2017 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Vyacheslav Makler, Christina L. Goldstein, Daniel Hoernschemeyer, Tomoko Tanaka. Chiari I malformation and syringomyelia in mucopolysaccharidosis type I (Hurler syndrome) treated with posterior fossa decompression: Case report and review of the literature. 26-May-2017;8:80
How to cite this URL: Vyacheslav Makler, Christina L. Goldstein, Daniel Hoernschemeyer, Tomoko Tanaka. Chiari I malformation and syringomyelia in mucopolysaccharidosis type I (Hurler syndrome) treated with posterior fossa decompression: Case report and review of the literature. 26-May-2017;8:80. Available from: http://surgicalneurologyint.com/surgicalint-articles/chiari-i-malformation-and-syringomyelia-in-mucopolysaccharidosis-type-i-hurler-syndrome-treated-with-posterior-fossa-decompression-case-report-and-review-of-the-literature/
Date of Submission
02-Dec-2016
Date of Acceptance
22-Feb-2017
Date of Web Publication
26-May-2017
Abstract
Background:Hurler Syndrome is the most severe phenotype of mucopolysaccharidosis type I. With bone marrow transplant and enzyme replacement therapy, the life expectancy of a child with Hurler syndrome has been extended, predisposing them to multiple musculoskeletal issues most commonly involving the spine.
Case Description:This is the case report of a 6-year-old male with Hurler syndrome who was diagnosed with Chiari I malformation and cervicothoracic syringomyelia on a preoperative magnetic resonance imaging (MRI) for his thoracolumbar kyphosis. This report details the successful management of a Chiari I malformation and syringomyelia with posterior fossa decompression in a child with Hurler syndrome.
Conclusion:Children born with MPS I can have complex spine issues that require surgical management. The most common orthopedic spinal condition for these patients, thoracolumbar kyphosis, requires evaluation with an MRI before performing surgery. This resulted in the diagnosis of a Chiari I malformation and syringomyelia in our patient with Hurler syndrome. This was successfully treated with decompression of the posterior fossa.
Keywords: Chiari I Malformation, Hurler syndrome, mucopolysaccharidosis type I, posterior fossa decompression, syrinx
INTRODUCTION
Mucopolysaccharidosis type I (MPSI) is an autosomal recessive lysosomal storage disease caused by deficient or absent activity of the α-L-iduronidase enzyme (IDUA), which catalyzes the degradation of the glycosaminoglycans (i.e., dermatan and heparan sulfates), the most severe form of which is Hurler syndrome (HS).[
CASE REPORT
The patient is a 6-year-old male who was referred to a pediatric clinic for an incidentally found CM-I and cervicothoracic syrinx [
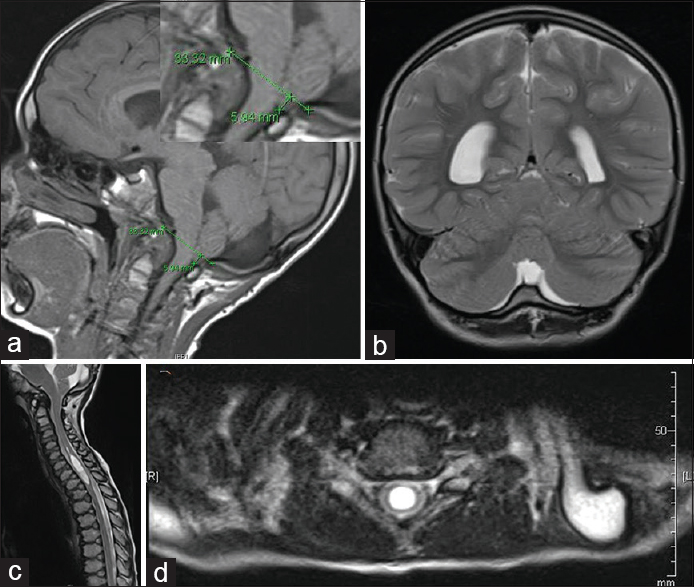
Figure 1
Preoperative MRI findings. (a) MRI brain, T1WI sagittal view. Demonstrates cerebellar ectopia, measuring approximately 5.9 mm with crowding within the foramen magnum. (b) MRI brain, T2WI coronal view. Demonstrates prominent retrocerebellar cystic space. The differential diagnosis includes major cisterna magna versus an arachnoid cyst. (c) MRI cervicothoracic spine, T2WI sagittal view. Demonstrates 8 mm syrinx from C5 to T1-2. (d) MRI cervicothoracic spine, T2WI axial view at the level of C6-7
On physical examination, the patient was noted to be alert and oriented with clear speech and normal cranial nerve function. Muscle strength was 5/5 in bilateral biceps, triceps, and deltoids, 4+/5 in hand grip and finger abduction. Deep tendon reflexes were grade 2/4 in the upper and lower extremities with a negative Hoffman's sign bilaterally, no ankle clonus, and a downgoing Babinski test bilaterally. The patient was unable to perform single-leg stance.
On August 4, 2015 the patient was taken to the operating room where he underwent a successful suboccipital craniectomy measuring 3 × 3 cm, C1 laminectomy, intradural exploration with coagulation of cerebellar tonsils, lysis of arachnoid adhesions, resection of thick posterior arachnoid membrane, and duraplasty measuring 3 × 3 cm utilizing Dura-Guard™ (Baxter Healthcare Corporation, Mountain Home, Arizona) and DuraSeal™ (Medtronic, Minneapolis, Minnesota). Intraoperatively, he was noted to have numerous arachnoid adhesions, an arachnoid cyst, and cerebellar tonsillar herniation to the level of C1 was confirmed. Postoperative course was unremarkable, and he was ultimately discharged home on postoperative day 3.
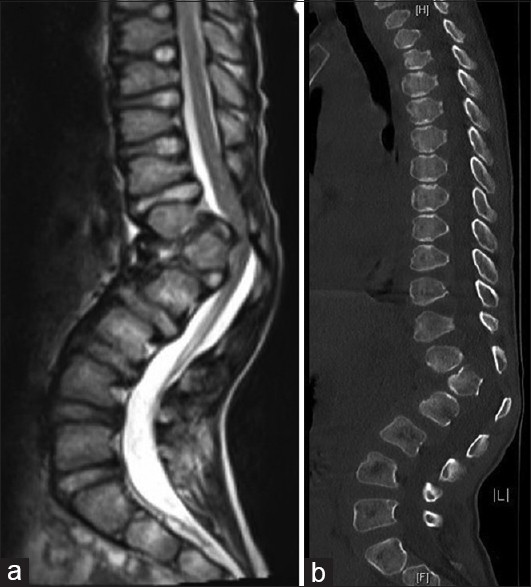
At his first 2-week postoperative follow-up, his mother reported more stability with his gait and improved balance. The patient returned to school. The patient's physical examination was unchanged from his preoperative exam except that he was now able to perform a single-leg stance. At his 3-month follow-up visit, his parents reported that he continued to improve. His daily headaches had resolved, and the MRI of the cervicothoracic spine revealed improvement in the size of the syrinx measuring 3.5 mm at its widest point [
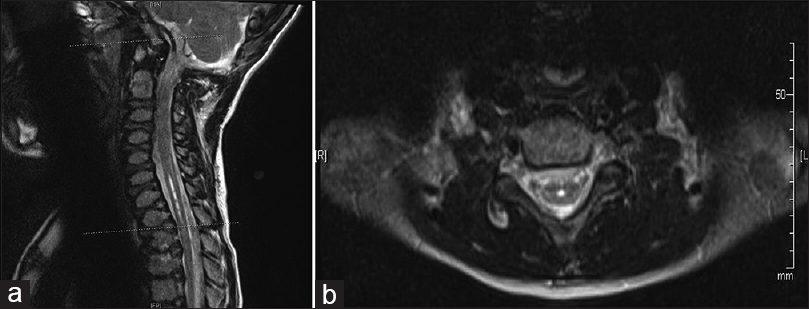
Six months after suboccipital craniectomy, the patient underwent an uncomplicated anterior release and posterior spinal fusion with correction of his thoracolumbar kyphosis. During the most recent 3-month follow-up visit, the patient was found to have complete resolution of his gibbus deformity and no changes with his neurologic exam. The MRI of the cervical and thoracic spine revealed a stable syrinx. This case report was approved by the University of Missouri Health Sciences Institutional Review Board, and an informed consent was obtained from the patient's parents.
DISCUSSION
In 1891, Hans Chiari documented three cases of congenital defects of the rhombencephalon, classified as type I, II, and III.[
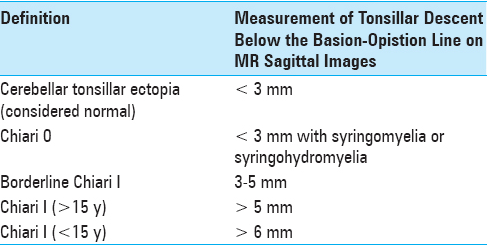
MPSI is an autosomal recessive lysosomal storage disease caused by mutation in the IDUA gene located on chromosome 4p16.3,[
Historically, MPSI has been divided into three clinical subtypes – Hurler (severe), Scheie (mild/attenuated), and Hurler–Scheie (intermediate).[
HS progresses rapidly from 6 to 24 months resulting in significant multiorgan dysfunction.[
Manara et al.,[
Hite et al.,[
In 2003, Zafeiriou et al.,[
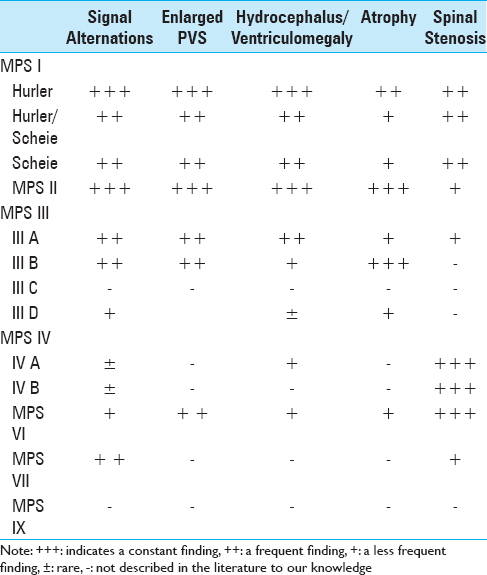
Tandon et al.,[
PFD has long been performed to relieve compression and restore normal CSF pathways at the craniocervical junction.[
CONCLUSION
MPSI is an autosomal recessive lysosomal storage disorder causing a chronic, progressive multiorgan disease by deficient or absent activity of the α-L-iduronidase enzyme, which catalyzes the degradation of glycosaminoglycans.[
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Aldenhoven M, Boelens JJ, de Koning TJ. The clinical outcome of Hurler syndrome after stem cell transplantation. Biol Blood Marrow Transplant. 2008. 14: 485-98
2. Baehner F, Schmiedeskamp C, Krummenauer F, Miebach E, Bajbouj M, Whybra C. Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis. 2005. 28: 1011-7
3. Barkovich AJ, Wippold FJ, Sherman JL, Citrin CM. Significance of cerebellar tonsillar position on MR. AJNR Am J Neuroradiol. 1986. 7: 795-9
4. Boor R, Miebach E, Bruhl K, Beck M. Abnormal somatosensory evoked potentials indicate compressive cervical myelopathy in mucopolysaccharidoses. Neuropediatrics. 2000. 31: 122-7
5. Campos D, Monaga M. Mucopolysaccharidosis type I: Current knowledge on its pathophysiological mechanisms. Metab Brain Dis. 2012. 27: 121-9
6. Chiari H. Concerning alterations in the cerebellum resulting from cerebral hydrocephalus. 1891. Pediatr Neurosci. 1987. 13: 3-8
7. Chotai S, Kshettry VR, Lamki T, Ammirati M. Surgical outcomes using wide suboccipital decompression for adult Chiari I malformation with and without syringomyelia. Clin Neurol Neurosurg. 2014. 120: 129-35
8. Coletti HY, Aldenhoven M, Yelin K, Poe MD, Kurtzberg J, Escolar ML. Long-term functional outcomes of children with hurler syndrome treated with unrelated umbilical cord blood transplantation. JIMD Rep. 2015. 20: 77-86
9. Guffon N, Souillet G, Maire I, Straczek J, Guibaud P. Follow-up of nine patients with Hurler syndrome after bone marrow transplantation. J Pediatr. 1998. 133: 119-25
10. Hite SH, Krivit W, Haines SJ, Whitley CB. Syringomyelia in mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome): Imaging findings following bone marrow transplantation. Pediatr Radiol. 1997. 27: 736-8
11. Jayarao M, Sohl K, Tanaka T. Chiari malformation I and autism spectrum disorder: An underrecognized coexistence. J Neurosurg Pediatr. 2015. 15: 96-100
12. Johnson BA, Dajnoki A, Bodamer OA. Diagnosing lysosomal storage disorders: Mucopolysaccharidosis type i. Curr Protoc Hum Genet. 2015. 84: 11-8
13. Malm G, Gustafsson B, Berglund G, Lindstrom M, Naess K, Borgstrom B. Outcome in six children with mucopolysaccharidosis type IH, Hurler syndrome, after haematopoietic stem cell transplantation (HSCT). Acta Paediatr. 2008. 97: 1108-12
14. Manara R, Concolino D, Rampazzo A, Zanetti A, Tomanin R, Faggin R. Chiari 1 malformation and holocord syringomyelia in hunter syndrome. JIMD Rep. 2014. 12: 31-5
15. McVige JW, Leonardo J. Imaging of Chiari type I malformation and syringohydromyelia. Neurol Clin. 2014. 32: 95-126
16. McVige JW, Leonardo J. Neuroimaging and the clinical manifestations of Chiari Malformation Type I (CMI). Curr Pain Headache Rep. 2015. 19: 18-
17. Moore D, Connock MJ, Wraith E, Lavery C. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J Rare Dis. 2008. 3: 24-
18. Peters C, Balthazor M, Shapiro EG, King RJ, Kollman C, Hegland JD. Outcome of unrelated donor bone marrow transplantation in 40 children with Hurler syndrome. Blood. 1996. 87: 4894-902
19. Rocque BG, George TM, Kestle J, Iskandar BJ. Treatment practices for Chiari malformation type I with syringomyelia: Results of a survey of the American Society of Pediatric Neurosurgeons. J Neurosurg Pediatr. 2011. 8: 430-7
20. Shapiro EG, Lockman LA, Balthazor M, Krivit W. Neuropsychological outcomes of several storage diseases with and without bone marrow transplantation. J Inherit Metab Dis. 1995. 18: 413-29
21. Souillet G, Guffon N, Maire I, Pujol M, Taylor P, Sevin F. Outcome of 27 patients with Hurler's syndrome transplanted from either related or unrelated haematopoietic stem cell sources. Bone Marrow Transplant. 2003. 31: 1105-17
22. Tandon V, Williamson JB, Cowie RA, Wraith JE. Spinal problems in mucopolysaccharidosis I (Hurler syndrome). J Bone Joint Surg Br. 1996. 78: 938-44
23. Wu T, Zhu Z, Jiang J, Zheng X, Sun X, Qian B. Syrinx resolution after posterior fossa decompression in patients with scoliosis secondary to Chiari malformation type I. Eur Spine J. 2012. 21: 1143-50
24. Xie D, Qiu Y, Sha S, Liu Z, Jiang L, Yan H. Syrinx resolution is correlated with the upward shifting of cerebellar tonsil following posterior fossa decompression in pediatric patients with Chiari malformation type I. Eur Spine J. 2015. 24: 155-61
25. Yang JS, Min HK, Oh HJ, Woo HI, Lee SY, Kim JW. A simple and rapid method based on liquid chromatography-tandem mass spectrometry for the measurement of alpha-L-iduronidase activity in dried blood spots: An application to mucopolysaccharidosis I (Hurler) screening. Ann Lab Med. 2015. 35: 41-9
26. Zafeiriou DI, Batzios SP. Brain and spinal MR imaging findings in mucopolysaccharidoses: A review. AJNR Am J Neuroradiol. 2013. 34: 5-13

