

- Praque, Czech Republic
- Theoretical Neurosciences Research, LLC, Ridgeland, Mississippi, USA
- Faculty of Health and Social Studies, Institute of Radiology, Toxicology and Civil Protection, University of South Bohemia Ceske Budejovice, Branisovska, Czech Republic
- Laboratory of Applied Hydrobiology, University of South Bohemia, Husova tř. 458/102, 370 05 České Budějovice, Czech Republic
Correspondence Address:
Russell L. Blaylock
Laboratory of Applied Hydrobiology, University of South Bohemia, Husova tř. 458/102, 370 05 České Budějovice, Czech Republic
DOI:10.4103/sni.sni_407_17
Copyright: © 2018 Surgical Neurology International This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.How to cite this article: Anna Strunecka, Russell L. Blaylock, Jiri Patocka, Otakar Strunecky. Immunoexcitotoxicity as the central mechanism of etiopathology and treatment of autism spectrum disorders: A possible role of fluoride and aluminum. 09-Apr-2018;9:74
How to cite this URL: Anna Strunecka, Russell L. Blaylock, Jiri Patocka, Otakar Strunecky. Immunoexcitotoxicity as the central mechanism of etiopathology and treatment of autism spectrum disorders: A possible role of fluoride and aluminum. 09-Apr-2018;9:74. Available from: http://surgicalneurologyint.com/?post_type=surgicalint_articles&p=8838
Date of Submission
01-Nov-2017
Date of Acceptance
07-Feb-2018
Date of Web Publication
09-Apr-2018
Abstract
Our review suggests that most autism spectrum disorder (ASD) risk factors are connected, either directly or indirectly, to immunoexcitotoxicity. Chronic brain inflammation is known to enhance the sensitivity of glutamate receptors and interfere with glutamate removal from the extraneuronal space, where it can trigger excitotoxicity over a prolonged period. Neuroscience studies have clearly shown that sequential systemic immune stimulation can activate the brain's immune system, microglia, and astrocytes, and that with initial immune stimulation, there occurs CNS microglial priming. Children are exposed to such sequential immune stimulation via a growing number of environmental excitotoxins, vaccines, and persistent viral infections. We demonstrate that fluoride and aluminum (Al3+) can exacerbate the pathological problems by worsening excitotoxicity and inflammation. While Al3+ appears among the key suspicious factors of ASD, fluoride is rarely recognized as a causative culprit. A long-term burden of these ubiquitous toxins has several health effects with a striking resemblance to the symptoms of ASD. In addition, their synergistic action in molecules of aluminofluoride complexes can affect cell signaling, neurodevelopment, and CNS functions at several times lower concentrations than either Al3+ or fluoride acting alone. Our review opens the door to a number of new treatment modes that naturally reduce excitotoxicity and microglial priming.
Keywords: Aluminofluoride complexes, aluminum, autism spectrum disorders, cytokines, fluoride, glutamatergic neurotransmission, immunoexcitotoxicity, microglial activation, neurodevelopment
INTRODUCTION
The mechanisms involved in autism spectrum disorder (ASD) etiopathology are many, but central to these disorders appears to be prolonged and repeated systemic immune activation and excitotoxicity. It has been generally accepted that chronic inflammation is the hallmark of many neurological and neurodegenerative diseases.[
The initial reaction to systemic inflammation may be microglial priming or full neurotoxic activation, depending on the intensity of the activating stimulus. It is known that a great number of conditions can lead to the priming/activation of microglia, such as stress, obesity, trauma, infections, sequential vaccination, hypoxia-ischemia, and exposure to certain neurotoxic metals, such as mercury, lead, and aluminum.
Microglia are involved in every aspect of neurodevelopment.[
There is growing evidence that glutamate and glutamate receptors (GluRs) are involved in neurodevelopment.[
In 1969, Olney demonstrated that exposure of specific types of neurons to high levels of glutamate caused a delayed death process that involved intense excitation of neuronal activity (excitotoxicity).[
In a previous article, we presented evidence that most heterogeneous symptoms of ASD have a common set of events closely connected with dysregulation of glutamatergic neurotransmission in the brain.[
In areas with fluoridated drinking water, we observe some symptoms of ASD, such as IQ deficits, sleep-pattern disturbance, inflammation, impaired ability of cognition, and learning and behavioral problems in some individuals. Prolonged exposure to fluoride in the prenatal and postnatal stages of development might have toxic effects on the development and metabolism of brain. Moreover, both fluoride and Al3+ interfere with a number of enzymes, resulting in a significant suppression of cellular energy production and oxidative stress.[
MICROGLIAL ACTIVATION IN BRAIN DEVELOPMENT AND AUTISM SPECTRUM DISORDER
Microglia and astrocytes are involved in every aspect of brain development, including synaptogenesis and refinement, synaptic pruning, neuron elimination, angiogenesis, as well as for maintenance, proliferation, differentiation, and migration of progenitor cells.[
Disturbance of microglial activation by immune stimulation during a critical stage can adversely affect synaptogenesis.[
NEURODEVELOPMENT AND MICROGLIA
Early in brain development a special glial scaffolding network derived from astrocytic cells, is formed called radial glial cells.[
Development and distribution of microglia in central nervous system
Microglia are derived from yolk sac progenitors that seed the brain around 4.5 weeks of gestation in the human brain and appear only when blood circulation develops.[
While amoeboid microglia migrate along radial glial cells and white matter in most mammals, in humans, microglia also invade and attach to blood vessels within the cortical parenchyma.[
Development of six-layered cortex, germinal zones, migration of progenitors to pial surface, and emergence of astrocytes and oligodendroglialcytes
The major germinal zones in the brain during embryogenesis include the ventricular zone (VZ) and the subventricular zone (SVZ) [
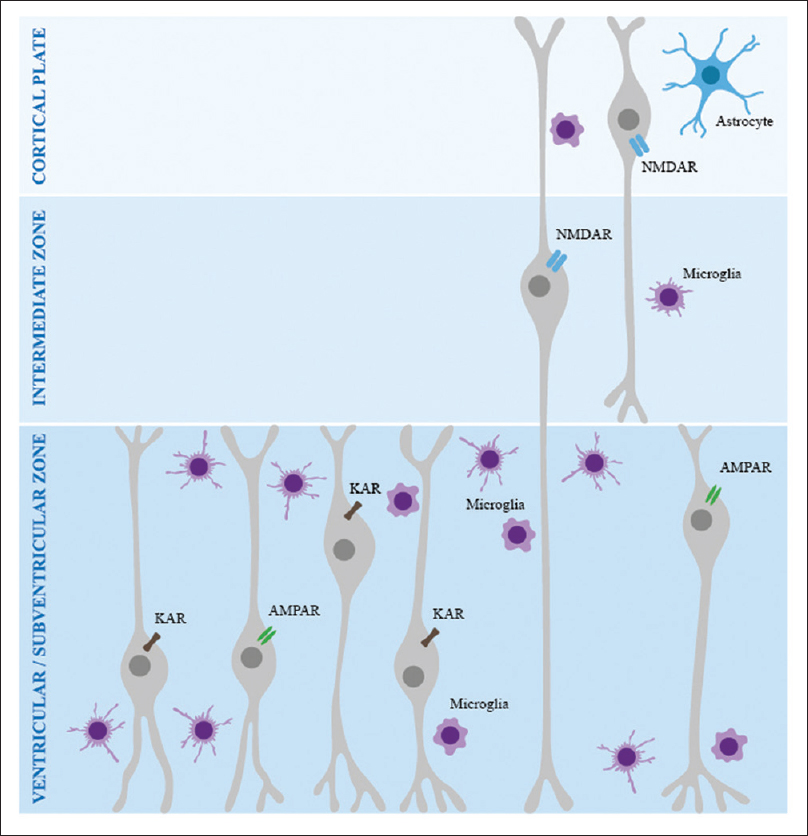
Figure 1
Microglia and Neurodevelopment. This illustration demonstrates how microglia plays a major role in all stages of neurodevelopment, by a programed release of cytokines and chemokines, a programmed release of glutamate and by active phagocytosis of excessive synaptic units, neurites and even whole cells. Acting at each of the germinal zones, VZ, IZ and CP, microglia controls cell proliferation, migration, angiogenesis, synaptogenesis, and dendritic and axonal development
Normal microglial “pruning” of neuropil, differences in microglial distribution by sex, and reason for early appearance in males of autism spectrum disorder
Cunningham et al. demonstrated that the size of the neuronal precursor pool was regulated by microglia, principally by phagocytosis.[
Females were shown to have a greater number of microglia than males, but this appeared later in development (P30–60). Interestingly, they also found that most microglia in the P4 males were of an activated (amoeboid) morphology, whereas the females at P30–60 were more often ramified [
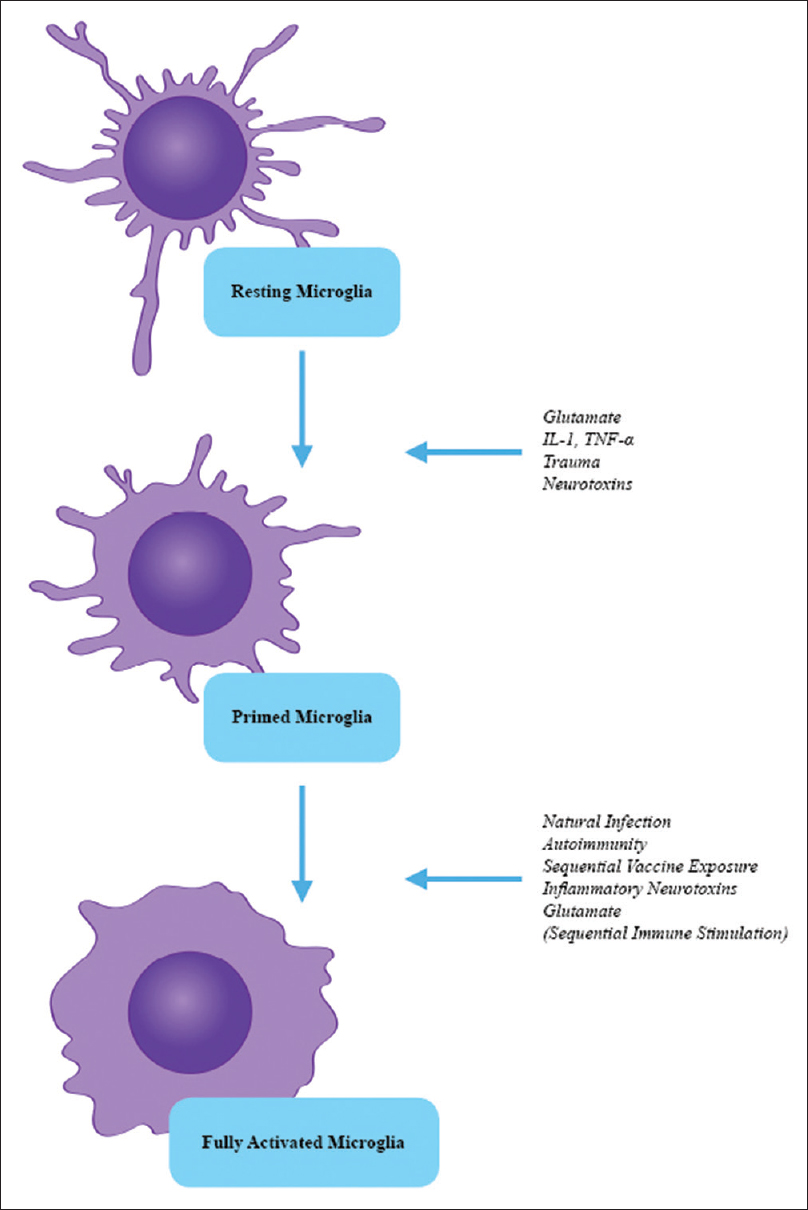
Figure 2
Microglial Priming and Full Activation by Sequential Immune Stimulation. Demonstrating the transition from a resting (ramified) microglial phenotype to a primed phenotype by an initial immune stimulus. Primed microglia have enhancement of cytokine generation, but no release of cytokines or excitotoxins. The second stimulus fully activates the microglia, resulting in a hyperintense immunoexcitotoxic reaction
Microglial population of different brain areas
Microglia colonizes different areas of the brain at significantly different rates.[
The neuroepithelial cells are the most primitive neural precursors, which give rise to all neurons, astrocytes, and microglia in the CNS. It is from the neural plate that we see the appearance of radial glia, which share properties with astrocytes. Importantly, these cells have specific glutamate transporters.[
New microglia are not derived from entering macrophages/monocytes, but rather are produced within the brain itself.[
Microglia concentration in cerebellum and autism spectrum disorder
Several studies have shown the cerebellum to be the most involved area of the brain in ASDs.[
Normal microglial pruning of synaptic connections; Abnormal microglia activation directed by compliment tags on synapses leads to abnormal neural development, and miswiring of brain in autism spectrum disorder; positron emission tomography scanning shows areas of microglia activation in autism spectrum disorder patients
Pruning is essential because during early neurodevelopment more synaptic connections are constructed than are ultimately needed by the mature brain. It has been shown that synaptic pruning is activity dependent, during which time large numbers of synapses are eliminated and the remaining ones are strengthened.[
Abnormal activation of microglia and/or the complement system leads to abnormal neural development, as seen in ASD.[
Suzuki et al. examined twenty men, aged 18–31 with confirmed autism versus matched controls using microglial activation PET scanning techniques utilizing the [11C]R-PK11195 tracer.[
What activates the microglia?
Microglia can be activated by peripheral autoantibodies, as are frequently seen in autistic patients.[
Reelen (a glycoprotein in brain development for neurla migration)
Reelin is a critical glycoprotein in brain development. Secreted from Cajal–Retzius cell in the marginal zone reelin plays an important role in neuronal migration in the prenatal and early postnatal brain.[
Maternal infections can lead to abnormal neural development as in autism spectrum disorder; Is the RELN gene a factor?
Human studies have shown that maternal infections can lead to several abnormalities of neurodevelopment, such as aberrant neural migration, decreased dendritic arborization, and abnormal cytoarchitecture in the adult, possibly related to abnormal reelin levels. Some authors suggested that reelin and RELN gene were significantly related with psychiatric disorders including ASD.[
CYTOKINES AND CHEMOKINES IN AUTISM SPECTRUM DISORDER AND NEURODEVELOPMENT; AUTISM SPECTRUM DISORDER AND IMMUNE DYSFUNCTIONS AND THE ROLE OF EXCITOTOXICITY IN AUTISM SPECTRUM DISORDER
It becomes evident that several cytokines and chemokines play essential roles in brain development regarding migration of progenitors, differentiation, maturation, dendrite arborization, and synaptogenesis. Significant alterations of these immune mediators during critical periods of brain development can also cause varying degrees of functional brain disruptions as well, resulting in abnormal connectivity and cytoarchitecture. Disturbances of these microglial-derived signaling molecules, such as systemic immune activation, would be expected to alter numerous brain pathways and functional systems.
Studies of autistic patients have confirmed the presence of a number of immune dysfunctions, such as neuroinflammation, brain directed autoantibodies, increased T-cell, NK cells, and macrophage responses.[
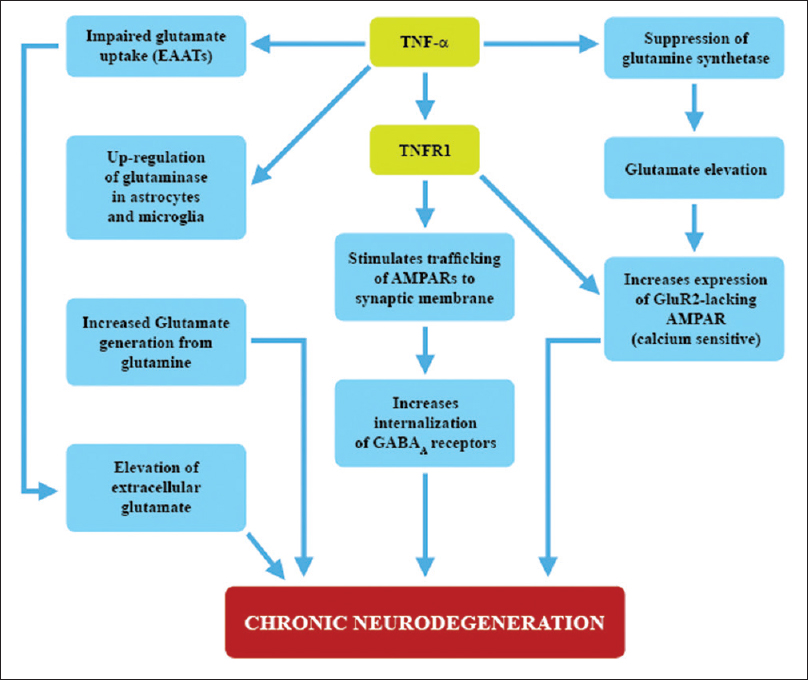
Figure 3
TNF-Alpha and Immunoexcitotoxicity This chart demonstrates how TNF-α, through one of its receptors (TNFR1) and via oxidative injury, is linked to enhanced excitotoxicity. This involves suppression of GS, trafficking GluA2-lacking AMPARs, upregulation of glutaminase, impaired glutamate transport and endocytosis of GABA receptors.
Using cerebellar granules cell cultures of individual populations of neurons, Oldeive and Doherty found that TNF-α had highly specific developmental effects on these neurons, depending on the embryonic stage of development.[
Together, these observations make a compelling case for a link between sequential systemic immune stimulation and neurobehavioral disorders such as the ASDs. Unfortunately, many studies and discussions have omitted or severely downplayed the role of other microglial components linked to immune destruction, such as excitotoxicity.
GLUTAMATE'S ROLE IN EXCITOTOXICITY AND AUTISM SPECTRUM DISORDER
In 1957, Lucas and Newhouse observed that mice exposed to high-dose monosodium glutamate (MSG) demonstrated extensive neuronal lesions in the inner layer of retinal neurons.[
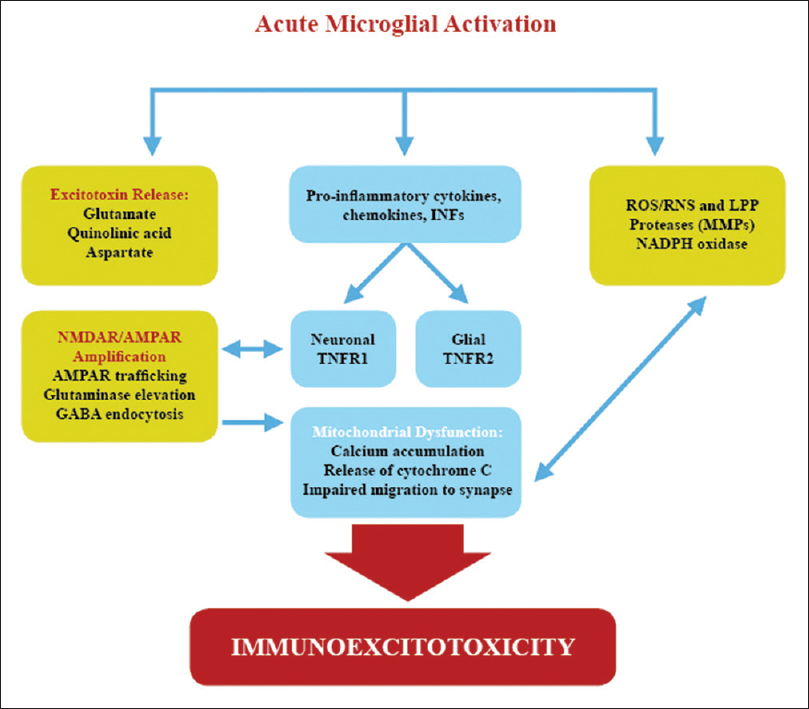
Figure 4
Aluminum and Aluminofluoride link to excitotoxicity. This diagram demonstrates stimulation of systemic immune activation and microglial activation within the brain by aluminum and fluoride as well as aluminofluoride. Al3+, F- and AlF4- all suppresses mitochondrial energy production and trigger ROS/RNS and LPP generation both directly and indirectly via microglial generated inflammatory cytokines, ROS/RNS, LPP and excitotoxicity. This can produce all the core features of ASD
THE GLUTAMATERGIC RECEPTOR SYSTEM – ITS LINK TO AUTISM SPECTRUM DISORDER AND ROLE IN EXCITOTOXICITY: AN OVERVIEW
Subsequent studies confirmed the status of glutamate as a neurotransmitter system and that it involved a number of specific types of receptors. Over 50% of the brain's neurotransmission is via glutamatergic neurotransmission and 90% of cortical neurons utilize glutamate as a neurotransmitter.[
The ionotropic GluRs are the N-methyl-D-aspartate receptors (NMDARs), amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (AMPAR), and kainate type receptors (KAR). NMDARs are linked to sodium, potassium, and Ca2+ channels. It is thought that the entry of excessive Ca2+ into the neuron plays a major role in the excitotoxic process by triggering cascades of destructive cellular signaling. Glutamate affinity is greater with NMDARs than AMPAR/KARs but AMPARs are faster conducting [
Glutamate receptor: NMDAR
Each of these receptor types is composed of a variable arrangement of subunits, which determines their biophysical and physiological properties. The NMDARs are composed of a tetrad arrangement of subtype receptor units–GluN1, GluN2A-D, and GluN3A and B (NR1, NR2A-D, and NR3A-B by the older classification system). Distribution of these variously composed NMDARs varies with their location in the brain. During development, there are significant changes in the composition of these subunits. In the mammalian brain, functional NMDARs require a GluN1 (NR1) subunit associated with one or more GluN2 (NR2) subunits. Magnesium (Mg2+) sites within NMDAR channels regulate receptor function, with Mg2+ playing a major role as a voltage-dependent channel blocker. With depolarization, the Mg2+ block is relieved, allowing the action potential to proceed. Sensitivity to Mg2+ blockade also varies with subunit composition.
Glutamate receptor: AMPAR
AMPARs are composed of GluA1-4 subunits (GluR1-4 by older nomenclature). The presence of the GluA2 (GluR2) subunit in the compositional structure of the AMPA receptor blocks Ca2+ entry into the neuron [
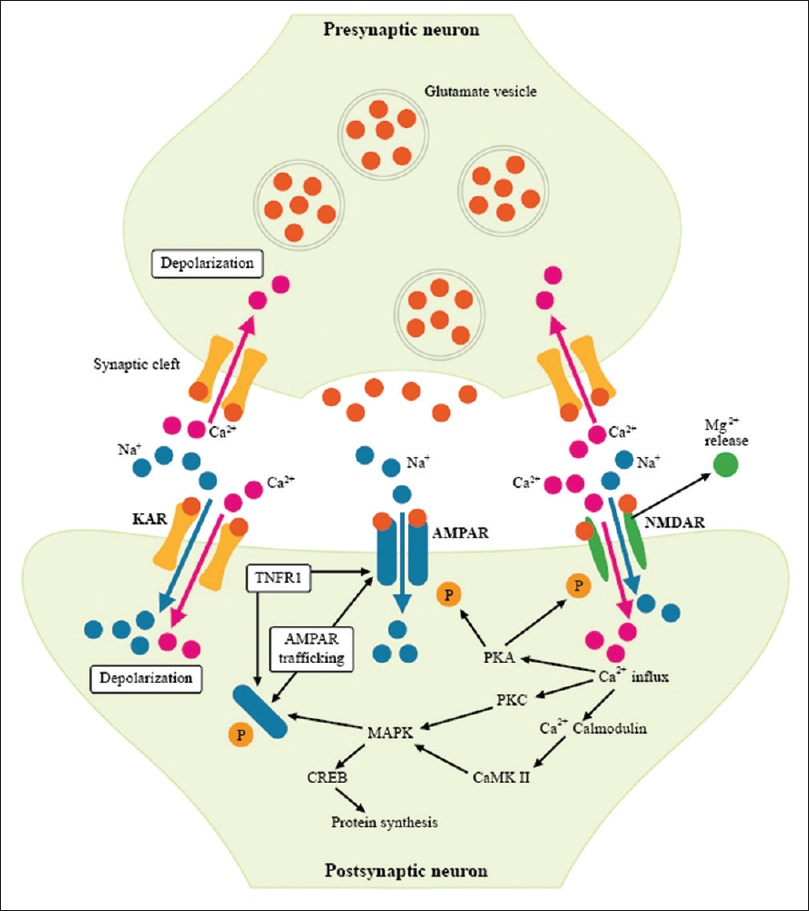
Figure 5
Synaptic Subunit Trafficking. Illustration of the pre- and post-synaptic units, demonstrating modulation of synaptic strength by trafficking of AMPA and NMDA subunits to the synaptic density. Trafficking of GluA2 (Glu2B)-lacking AMPARs, converts the AMPAR into a calcium permeable receptor, greatly increasing its excitatory strength. Presence of the GluA2 receptor within the AMPAR blocks calcium permeability. Inflammation converts AMPARs to the calcium permeable form
Glutamate receptors: Metabotropic receptors (mGluRs)
In addition to the ionotropic GluRs, researchers have identified three major classes of mGluRs with eight subunit types. These receptors operate through G-protein-coupled receptors (GPCRs) via seven transmembrane domains, with an extracellular N-terminal and intracellular COOH terminal domain. Group I mGluRs contain two subtype receptors, mGluR1 and mGluR5, which activate phospholipase C (PLC), producing inositol 1,4,5-trisphosphate, and diacylglycerol (DAG) as second messengers [
The mGluRs also vary in location, which affects their physiological effect. Some are close to the postsynaptic density and others occur on the presynaptic axon.[
GLUTAMATE AND GLUTAMATE RECEPTORS IN NEURODEVELOPMENT
Glutamate receptors (GluRs) are expressed very early in development and play a vital role in most aspects of neurodevelopment, including brain wiring, synaptogenesis, progenitor migration, differentiation, and maturation of cells.[
Within the intermediate germinal zone, a site of precursor cell proliferation, and the cortical plate, where neuronal differentiation occurs following radial migration of the precursors, we see further changes with developmental progression [
Furuta and Martin examined sheep neocortex during cortical lamination, specifically for expression of AMPARs (GluR1, GluR2/3 and GluR4), KARs (GluR6/GluR7), and mGluR5.[
Smith and Thompson, examining the visual cortex of ferrets, found that NMDARs and non-NMDARs follow quite distinct developmental patterns.[
Similar patterns of expression for the various subunits have been confirmed in the developing human brain as well.[
Sodium-dependent glutamate transporters have also been shown to play a major role in neurodevelopment, primarily by controlling glutamate levels, and hence, GluR activation during developmental stages. In humans, there have been five identified glutamate transporters, EAAT1-5. In animal species, GLAST represents EAAT1 and GLT-1, EAAT2. EAAT1 and EAAT2 are present on the cell membranes of microglia, astrocytes, and oligodendrocytes. EAAT1 (GLAST) is the major glutamate transporter in the cerebellum, being mostly expressed in astrocytes.[
Furuta et al. found that in the rat, GLAST levels were low prenatally in the forebrain, but were high in Bergmann glia in the cerebellum early postnatally.[
GROWTH CONES, GLUTAMATE RECEPTORS, AND CALCIUM GUIDANCE
Growth cones are specialized structures located at the tip of developing axons that act as guidance mechanisms for successful union with synaptic partners. Located at the leading edge of the growth cone are special structures called filopodia and lamellipodia, which guide the axon to its destination among an enormously complex array of possibilities. A number of studies have identified GluRs, in particular NMDARs, within the membrane of growth cones.[
Activation of the growth cone NMDARs produce calcium (Ca2+) oscillations that act as guidance signals for axon movement, which includes neuron migration, neurite outgrowth, motility, axon turning, and activation of intracellular signaling pathways involving Rho GTPases, all of which lead to brain architectural construction.[
Disruption of cortical column cytoarchitecture and connectivity is characteristic of ASDs. For example, Damarla et al. examined high functional autistics using a combination of behavioral testing, functional MRIs, functional connectivity, and corpus callosum morphometric methological tools, and found that autistics had lower functional connectivity between higher order working memory/executive areas and visuospatial regions (between frontal and parietal-occipital regions).[
Underconnectivity, with severe changes in many brain structures, has also been shown for the cerebellum, an area of the brain most affected in autism.[
Synaptogenesis and synaptic pruning within the brain follows a programmed timeline that is specific for individual areas of the brain. A recent study found that synaptic density begins to decline at puberty and is completed during adolescence, within the prefrontal cortex. Substantial elimination of synaptic spines continues beyond adolescence, well into the third decade.[
Glutamate receptors and the cerebellum in autism spectrum disorder
Within the cerebellum, it has been shown that GluA2 receptors, an AMPAR subunit that reduces Ca2+ influx, is required for normal development of Purkinje cell dendrites.[
Glutamate receptors and synapse formation
Presynaptic NMDARs also play a major role in synapse formation, and stabilization. GluN2B (NR2B) is essential for neural pathway construction and is widely expressed in the brain during neurodevelopment.[
Glutamate receptors: Metabotropic GluRs and neurodevelopment
Metabotropic GluRs have also been shown to play a major role in neurodevelopment.[
Glutamate receptors and autism spectrum disorder; A short neurobiochemical summary
Together, these studies strongly suggest that glutamate and its receptors are playing a major role in the progressive elaboration of the architecture of the developing brain and that a number of internal and external environmental conditions can alter both glutamate levels and GluR physiology, resulting in varying degrees of abnormal brain development.
IMMUNOEXCITOTOXICITY IN BRAIN DEVELOPMENT AND AUTISM SPECTRUM DISORDER
A growing number of studies are defining an interrelationship between the immune system and excitatory neurotransmission. This includes alteration of glutamate transporters as well as direction of glutamate transport, trafficking of Ca2+-permeable AMPARs and NMDARs, elevation of glutaminase activity, and suppression of mitochondrial function with alteration of mitochondrial energy generation, thus enhancing excitotoxicity [
FROM ALTERATION OF THE IMMUNE RESPONSE TO GLUTAMATE RECEPTORS TO EXCITOTOXICITY WITH EXCESS GLUTAMATE AS A NEUROTRANSMITTER
One of the principle control systems of extraneuronal levels of glutamate is accomplished by a series of five glutamate transport proteins (EAAT1-5) discussed above. Both inflammatory cytokines and excitotoxins, alter the redox status of EAATs, making them dysfunctional, thus allowing the accumulation of extracellular glutamate and possible excitotoxicity. Extraneuronal glutamate can also be altered by another system called the cystine/glutamate antiporter Xc-. Under normal conditions, this system exchanges extracellular cystine for intracellular glutamate, thus raising extracellular glutamate levels, which are then quickly lowered by EAATs [
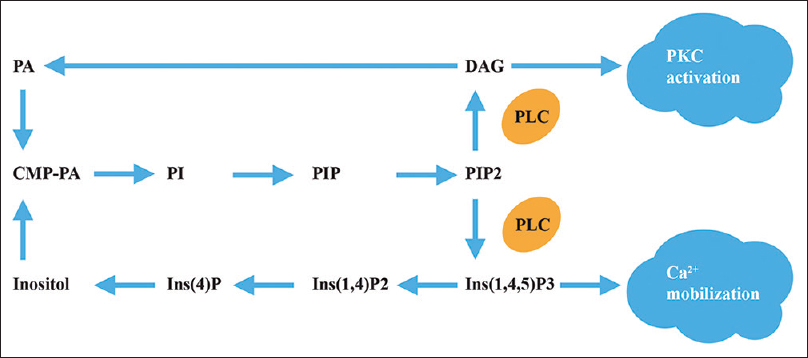
Figure 6
A simplified scheme of the phosphoinositide signaling system. Phosphatidylinositol 4,5-bisphosphate (PIP2) in the plasma membrane is hydrolyzed by phospholipase C (PLC) after GPCR activation and yields inositol 1,4,5-trisphosphate (Ins(1,4,5)P3) and diacylglycerol (DAG). Both products of this hydrolysis have a second messenger role. Ins(1,4,5)P3 binds to a receptor in membranes of endoplasmic reticulum, which results in a release of Ca2+ into the cytosol. DAG activates protein kinase C (PKC). The coupling between the GPCR and PLC is mediated by G proteins. PA – phosphatidic acid, PI – phosphatidylinositol.
REPEATED IMMUNE ACTIVATION LEADS TO MICROGLIAL ACTIVATION, EXCESS GLUTAMATE, AND NEUROPATHOLOGICAL CHANGES IN THE NEUROPIL THAT CAN BE BLOCKED BY GLUTAMATE RECEPTOR BLOCKERS
Prolonged and repeated systemic immune activation appears to be central to ASD etiopathology. With subsequent, especially closely spaced immune stimulation, full activation of the overactive primed microglia occurs, leading to progressive pathological changes in the developing brain. This is especially so if the episodes of systemic immune stimulation are closely spaced together.[
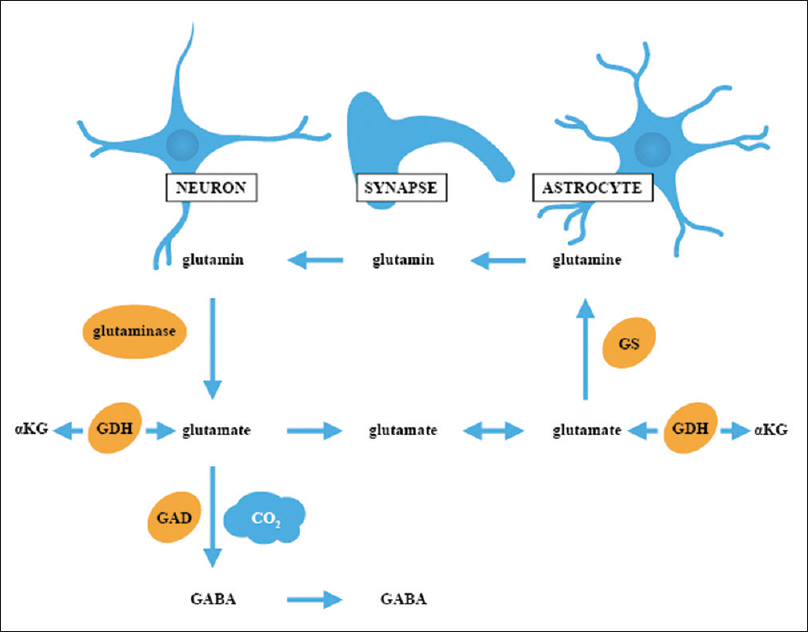
INTEGRATION OF IMMUNE DYSFUNCTION, GLUTAMATE RECEPTOR CHANGES, MICROGLIAL ACTIVATION, AND DISRUPTION OF NEURODEVELOPMENTAL MILESTONES LEADING TO PATHOLOGIC CHANGES IN AUTISM SPECTRUM DISORDER
In most studies, the target was neurodegeneration, yet the effects of elevation of glutamate brain levels during neurodevelopment can occur at concentrations lower than needed to cause neurodegeneration. This means that microglial activation alone, as long as it is associated with an elevation of glutamate levels, holds the possibility of disrupting neurodevelopmental milestones, particularly if occurring at periods when glutamate levels should fall. This would especially be of concern should inflammation be prolonged and associated glutamate levels were constantly elevated as well, as we see in ASDs.[
Of particular interest is the inflammation-induced enhanced trafficking of GluRs and alteration of glutamate subunits, as well as inflammatory cytokine-stimulated endocytosis of GABA receptors, which would shift the balance in the brain toward excitotoxicity. The interaction between immune mediators and GluRs triggers a neurotoxic reaction, which involves the generation of high levels of ROS and RNS, along with LPP. These neurotoxic compounds have been shown to inhibit glutamate transport proteins (EAATs), inhibit metabolic glutamate-clearing enzymes, such as glutamine synthetase (GS), glutamate dehydrogenase (GDH) and glutamic acid dehydrogenase (GAD), as well as interfere with mitochondrial energy generation [
Interestingly, maternal inflammation has also been associated with a significant increase in glutaminase, an increase in NMDAR subunit GluN2 (NR2) expression and impairment of GLT-1 function, all things that trigger excitotoxicity.[
The ability of immune cytokines and other immune mediators of inflammation, to enhance microglial priming/activation and to magnify the excitotoxic potential of GluRs, indicate that the two processes operate together, both physiologically and pathologically.
THE ROLE OF FLUORIDE AND ALUMINUM IN ETIOPATHOLOGY OF AUTISM SPECTRUM DISORDER
A considerable amount of scientific evidence demonstrates that fluoride and Al3+ can harm a great number of physiological functions, including critical regulatory enzymes, mitochondrial energy production, neurotransmitter and endocrine signaling, brain development, higher CNS functions, and immune system dysfunctions.[
FLUORIDE AS A NEUROTOXIN IS IN ENVIRONMENTAL EXCESS IN GROWING YOUNG CHILDREN
Fluoride is a ubiquitous compound found in drinking water (both naturally and added) in many soils, incorporated within edible plant components, and is considered a natural compound. Millions of people live in endemic areas with high concentrations of fluoride in groundwater and in the biosphere. Fluoride exposure is common in fetuses, newborns, and small children. The United States Environmental Protection Agency (EPA) has done both a dose-response analysis and a relative source contribution analysis.[
EXCESS FLUORIDE AND NEURODEVELOPMENTAL DEFECTS
Fluoride is not an essential element for human growth and development. Prolonged exposure to fluoride in the prenatal and postnatal stages of development might have toxic effects on the development and metabolism of brain. In fluoridated areas, we observe some core symptoms of ASD in some individuals. These include IQ deficits, hypocalcemia, hypomagnesemia, hypothyroidism, sleep-pattern disturbance, inflammation, impaired ability of cognition, learning, and behavioral problems. Blaylock was the first to suggest that excitotoxicity may be a central mechanism of fluoride toxicity.[
ALUMINUM AS A NERUOTOXIN
There are a number of Al3+ sources, such as the drinking water, dietary substances, cosmetics, and the widespread use of Al3+ in medicine, namely in vaccines. Many investigations show that Al3+ can elicit impairment of development and immunity; that it acts as a hormonal disruptor, a neurotoxin, and elicits intense and prolonged activation of brain inflammation. Al3+ toxicity in humans, especially as regards the CNS, has been studied and discussed by several authors [e.g. 23,171,256,271. It is not surprising that Al3+ appears among the environmental toxins, which can participate in the etio-pathology of ASD.[
INCREASE IN EXPOSURE TO ALUMINUM; RELATIONSHIP BETWEEN ALUMINUM EXPOSURE AND INCREASE IN AUTISM SPECTRUM DISORDER
Among the suspected environmental toxins surveyed, Al3+ and Al3+-adjuvants have increasingly correlated positively to the rise in ASD.[
Widespread mobilization of Al3+ into our biosphere may place all of humankind at a heightened inflammatory status thereby increasing general risk for the development of ASD and other progressive neurodegenerative disorders associated with an inflammatory component.[
ALUMINUM AND FLUORIDE EXPOSURE INDUCES MICROGLIA ACTIVATION, INCREASES GLUTAMATE BLOOD AND BRAIN LEVELS AND ABNORMALITY IN THE DEVELOPING BRAIN AND NEURODEGENERATION
Recent studies have also shown that both fluoride and Al3+, as well as AlFx can induced microglial, astrocyte and B-cell activation, with resulting increases in blood and brain ROS, RNS, and LPP.[
METABOLIC AND DEVELOPMENTAL EFFECTS OF FLUORIDE
The highly electronegative fluoride ion with the same size and the same valence orbital as oxygen became a useful laboratory tool in our understanding of the biochemical and biophysical mechanisms of enzyme catalysis underlying biological processes such as metabolism and signal transduction.[
Almost 50% of children with ASD may display peripheral markers of disturbances of mitochondrial energy metabolism.[
Several researchers have reported evidence of mitochondrial dysfunction in ASD brain samples compared to controls.[
Recently, Delhey et al. measured mitochondrial enzyme activity on the one of the largest cohorts of individuals with ASD studied to date with concurrent measurement of symptoms in a subset and found that children with ASD demonstrated significantly greater variation in mitochondrial activity compared to controls.[
It has also been demonstrated that suppression of energy generation significantly increases sensitivity to excitotoxicity.[
One of the best documented biochemical changes in ASD is a decrease in cellular glutathione (GSH), a major intracellular antioxidant, and an increase in oxidized glutathione (GSSG), resulting in a reduction in the ratio of reduced (active) GSH to inactive GSSG [
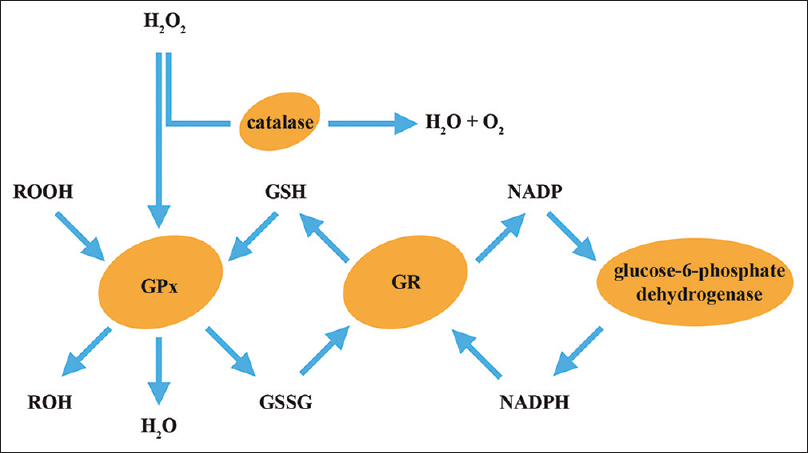
Figure 9
The GSH redox system. Hydrogen peroxide (H2O2), the immediate precursor of hydroxyl radical, is metabolized via the action of catalase and glutathione peroxidase (GPx). GPx also metabolizes reactive hydroperoxides (ROOH) and oxidizes reduced glutathione (GSH) to its disulfide form (GSSH), which is recycled back to GSH by the action of glutathione reductase (GR). A cofactor for GR is NADPH, which is supplied by the action of glucose-6-phosphate dehydrogenase
GSH, a radical scavenger, is converted to GSSG through glutathione peroxidase (GPx), and converted back to GSH by glutathione reductase (GR). GSH can detoxify hydrogen peroxide (H2O2), preventing the formation of hydroxyl radicals [
The reduced/oxidized GSH-redox equilibrium regulates a pleiotropic range of functions that includes ROS/RNS scavenging, detoxification, maintaining cell membrane integrity, signal transduction, and apoptosis. Under normal physiologic conditions, glutathione reductase activity is sufficient to maintain the high GSH/GSSG ratio. Excessive intracellular oxidative stress could result in GSSG export to the plasma. Therefore, an increase in plasma GSSH is a strong indicator of intracellular oxidative stress, which may have functional consequences such as increased mitochondrial superoxide production and a chronic inflammatory response.[
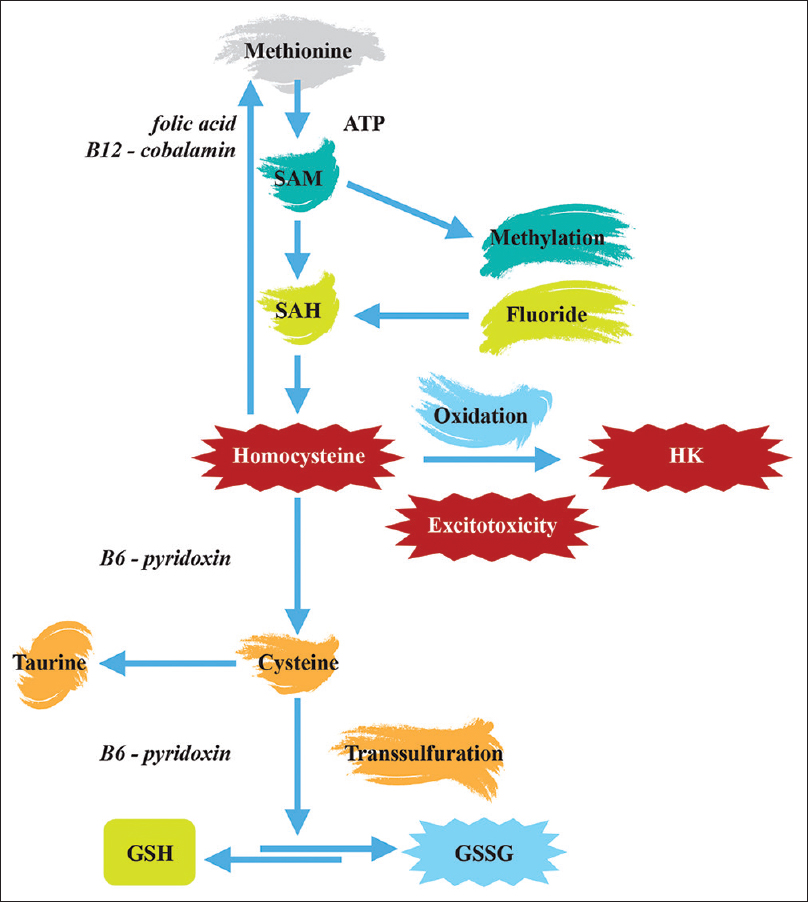
GSH and GSSG levels are significantly changed in autistic individuals, mainly as the consequence of disturbances in processes of transmethylation/transsulfuration. Plasma concentrations of methionine, S-adenosylmethionine (SAM), S-adenosylhomocysteine (SAH), adenosine, homocysteine, cystathionine, cysteine, GSH and GSSG have been repeatedly measured in children with autism and found to be disturbed[
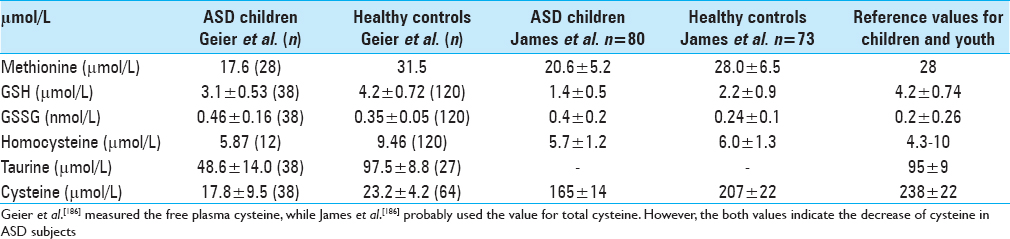
Highly significant (P ≤ 0.001) decrease of SAM and SAH was also observed in subjects with ASD.[
Fluoride can contribute to disturbances in GSH homeostasis because it enters the cascade and attaches to the SAH. Fluoride has been identified as an inhibitor of SAH hydrolase.[
Oxidative stress, which can stimulate microglial activation and immunoexcitotoxicity, may lead to neurodevelopmental abnormalities in children by affecting numerous cellular processes, especially via cell signaling and mitochondrial dysfunction.
THE EFFECTS OF FLUORIDE ON THE BRAIN AND NEURODEVELOPMENT
Numerous animal studies have been published, which have raised a level of concern about the impact of increasing fluoride exposure on the brain of individuals with autism.[
Human studies validating animal observations of fluoride toxicity
Two studies provided evidence that peripheral markers of oxidative stress observed in plasma and immune cells of subjects with ASD are similarly changed in several brain regions. Chauhan et al. found that GSH levels were significantly decreased by 34.2 and 44.6%, with a concomitant increase in the levels of GSSG by 38.2 and 45.5%, respectively, in the cerebellum and temporal cortex from patients with autism compared to the control group.[
Rose et al. examined frozen samples from the cerebellum (n = 15) and Brodmann area 22 (BA-22) of the temporal cortex (n = 12) from individuals with autism and unaffected controls. GSH and GSH/GSSG were significantly decreased in both the autistic cerebellum and BA-22.[
However, unlike systemic cells, brain neurons depend on the transfer of astrocytic generated GSH, which is produced by the glutamate/cystine antiporter Xc-. The glutamate/cystine antiporter Xc- is an exchange system involving an exchange of intracellular glutamate for extracellular cystine. Intracellular cystine is broken down into cysteine, which is metabolized to GSH. Cysteine is the rate-limiting substrate for GSH and, along with cystine, it also forms a key redox couple on its own. That is, they act to reduce oxidative stress. High levels of extraneuronal glutamate inhibit the exchange and can lower brain GSH.[
Human studies on impaired neural development found in brains of aborted fetuses in an endemic flourosis area of China by a pathological study and by a IQ assessment study
Based on the research from China, the fetal brain is highly susceptible to fluoride poisoning.[
Meta-analyses studies of intelligence in children with high fluoride exposure
Tang et al. published a meta-analysis of 16 studies carried out in China between 1998 and 2008 evaluating the influence of fluoride levels on the IQ of children.[
Findings from meta-analyses of 27 studies published over 22 years suggest an inverse association between high fluoride exposure and children's intelligence.[
More human studies on the inverse association of fluoride exposure and IQ
The team of Fluoride Action Network summarized 57 human studies; 50 of which found that elevated fluoride exposure is associated with reduced IQ and only 7 studies found no such association (
The results from several geographic regions support the view that fluoride may be a developmental neurotoxicant that affects brain development at exposures much below those that can cause toxicity in adults. Serum fluoride concentrations associated with high intakes from drinking water may exceed 1 mg/L (50 μmol/L) – more than 1,000 times the levels of some other neurotoxicants that cause neurodevelopmental damage.[
FLUORIDE AND ENDOCRINE DISRUPTIONS
Fluoride and thyroid dysfunction
Disturbance of thyroid hormone production has been found in correlation with lowered IQ in children. The investigations from India demonstrated that the thyroid gland appears to be very sensitive tissue in the body in relation to fluoride burden. Susheela et al. compared the production of free thyroid hormones (FT3/FT4) and thyroid-stimulating hormone (TSH) of 90 children living in fluoride endemic, non-iodine deficient areas of Delhi, India, along with 21 children from non-endemic areas.[
Thyroid deficiency leads to brain damage in autism spectrum disorder
Thyroid hormones are essential for brain maturation and for brain function throughout life. Thyroid hormone deficiencies even for short periods, may lead to irreversible brain damage, the consequences of which depend on the specific timing of onset and duration of thyroid hormone deficiency.
Thyroid dysfunction is frequently found in children with ASD.[
FLUORIDE ACCUMULATION IN THE PINEAL GLAND; MELATONIN DEFICIENCY AND AUTISM SPECTRUM DISORDER
Some symptoms of ASD, such as the sleep problems and the early onset of puberty, suggest abnormalities in melatonin physiology and dysfunctions of the pineal gland.[
Decreased levels of melatonin in blood or urine have been reported as very frequent features in individuals with ASD compared to typically developing controls.[
FLUORIDE AND NEUROINFLAMMATION: EVIDENCE FROM ANIMAL STUDIES
Few have examined the possibility of direct microglial activation by fluoride, despite that Shivarajashankara with colleagues reported in 2003 increased levels of ROS, RNS, and LPP in the blood of experimental animals treated with fluoride.[
In astrocytes, fluoride activates phospholipase C (PLC) and increases intracellular Ca2+ level. Once activated by fluoride, microglia secrete large concentrations of IL-1ß and TNF-α, which can then recruit more microglial activation in a vicious cycle that ultimately leads to neurodegeneration. Likewise, excitotoxins can activate microglia and stimulate release of inflammatory cytokines and additional glutamate.[
The abovementioned experiments suggest that the toxic effects of fluoride on the CNS may be attributed to the release of inflammatory cytokines and ROS, but fluoride concentration used in animal trials is very high. In humans, stimulation of the immune system with fluoride is not generally discussed among fluoride's adverse effects.
ALUMINUM AS ENVIRONMENTAL TOXIC SUBSTANCE
Al3+ has, until relatively recently, existed in forms not generally available to living organisms, and was therefore regarded as nontoxic. We underscore the remarkable and ongoing liberation of Al3+ into the biosphere, resulting in its increased bioavailability to biological systems. The most significant factor driving complacency about the potential dangers of Al3+ is its omnipresence in modern life. It is likely that Al3+ is present in every physical and chemical compartment in the human body.[
However, Al3+ is a non-essential element and it has no biological function in humans.
Sources of aluminum; entry into body
There are a number of ways in which humans are exposed to Al3+, such as through the skin, the lung, the nose, gastrointestinal system and of course, via intramuscular vaccination. Other major contributors include Al3+ used in medicines: dialysis solutions, parental and intravenous nutrition solutions used in pediatrics. Vaccines, allergy skin tests, human serum albumin, baby skin creams, baby diaper wipes and antacids, which are frequently given to infants, are extremely high in Al3+.
The primary natural source of Al3+ is food, which provides approximately 16–100 fold more Al3+ to systemic circulation than drinking water. While Al3+ content of breast milk is very small (about 20 μg/L), a recent review of 30 infant formulas found that all contained Al3+ and a number had concentrations far in excess of safety standards for oral consumption.[
Aluminum as an activation agent of microglial-induced immunoexcitotoxicity
As a powerful immune activating element, Al3+ can act both as the priming agent and the activation agent of microglial-induced immunoexcitotoxicity. This can occur with environmental exposure to excess Al3+, as well as injection by vaccination. The major difference is that ingested Al3+ is very poorly absorbed, whereas injected Al3+ nanoaggregates with antibodies is completely absorbed and distributed throughout the body, including the brain.[
Aluminum-induced neurodegeneration
With the demonstrated widespread accumulation of Al3+ in the brain and CSF following Al3+ exposure, all the conditions for an intense and prolonged activation of brain inflammation are present. Al3+ can activate microglia leading to secretion of TNF-α, IL-6 and cytokine-inducible NO synthase, and the development of neuroinflammatory ROS/RNS.[
In animal models, aluminum leads to increased glutamate, potentiates damaging redox activity, displaces metal ions with strong binding capacity in cellular reactions, alters enzymes of oxidative metabolism in mitochondria, enters the brain, is active in developing brains, which could contribute to neurodegeneration.
Al3+ also induces significant alterations of glutamate/glutamine recycling within astrocytes leading to increased glutamine to glutamate conversion coupled with increased uptake of glutamate (see section 3).[
Al3+ alters the activity of several enzymes of oxidative metabolism in mitochondria, such as a significant decrease in the activity of cytochrome C oxidase, NADH and succinate dehydrogenase.[
In a study of the fate of injected Al3+-adjuvants into muscle, researchers found that the Al3+ particles were taken up mostly by microglia.[
Al3+ has no known beneficial physiological action in the human body. Al3+ induced events likewise include oxidative stress, disruptions of energy metabolism, inflammation, glutamate excitotoxicity and effects on Ca2+ homeostasis, and therefore might participate in etio-pathology of ASD.
THE ROLE OF ALUMINOFLUORIDE COMPLEXES IN ETIOPATHOLOGY OF AUTISM SPECTRUM DISORDER
Binding of aluminum to fluoride
The synergistic action of fluoride and Al3+ has an important implication for pathological injury. Fluorine is the most chemically reactive nonmetal and the most electronegative element. Al3+ binds fluoride anion more strongly than 60 other metal ions tested by Martin.[
Aluminofluoride as an analogue of phosphate groups in biochemical reactions
Under physiological conditions and even with micromolar concentrations of Al3+, these two atoms react to form AlF4−, a molecule whose shape and physical properties closely resemble those of the phosphate anion, PO43-. AlF4− has been widely used as an analogue of phosphate groups to study phosphoryl transfer reactions and heterotrimeric G proteins involvement. Phosphoryl-transfer reactions are involved in processes such as regulation of cell metabolism, energy transduction, cytoskeletal protein assembly, regulation of cell differentiation and growth, aging, and apoptosis. Numerous laboratory studies demonstrated that AlF4− interacts with all known G proteins and this feature has been exploited to help researchers understand phosphate-dependent reactions in signaling cascades.[
Aluminofluoride binding to ADP and GDP b-phosphate and effects on protein conformation, interference in other signaling cascades
AlF4− binds ionically to the terminal oxygen of ADP or GDP β-phosphate. ADP or GDP could therefore form a complex with AlF4− that imitates ATP or GTP in its effect on protein conformation. This effect causes a structural change that locks the site and prevents the release of the γ-phosphate. Several authors have presented evidence that AlF4− interferes with regulatory GTP hydrolases, which play an initiating role in regulation of effector enzymes.[
Aluminofluoride and its influence on the immune system
Fluoride in the presence of micromolar Al3+ affects the function of lymphocytes and cells of the immune system, ion transport, Ca2+ influx and mobilization, protein phosphorylation, processes of neurotransmission, growth and differentiation, cytoskeletal proteins, and many other processes. These effects are not surprising when considering the extensive role of G proteins and GPCRs in the cell physiology. Approximately 800 GPCRs are encoded in the human genome. Physiological agonists include neurotransmitters and hormones, such as glutamate, dopamine, serotonin, melatonin, acetylcholine, TSH, neuropeptides, and excitatory amino acids.
Gs proteins stimulate adenylyl cyclase (AC) to produce intracellular cyclic adenosine monophosphate (cAMP), whereas Gi proteins inhibit the same process. Members of the cAMP-dependent second-messenger pathways participate in the regulation of cellular growth and differentiation and are also implicated in a variety of embryonic stages including brain development. cAMP is one of the key factors for neuronal outgrowth, plasticity, and regeneration.
Animal studies on aluminum leading to autism spectrum disorder; suppression of melatonim
Notably, the autistic-like behaviors were observed in an animal model of adenylyl cyclase type 5 (AC5) knockout (KO) mice. Kim et al., reported that loss of AC5 in the dorsal striatum produces increased repetitive behaviors and sociability deficits.[
In animal models aluminofluoride effects metabolic processes governing Ca2+ regulation that can affect excitotoxicity, can lead to neurodegeneration; low dose fluoride enhances uptake of aluminum, fluoride in tap water likely fed to infants; Aluminofluoride complex can lead to neurodegeneration
Many of metabolic and physiological processes are affected by alterations in intracellular Ca2+ levels. Gαq mediate phospholipase C (PLC)-dependent phosphoinositide hydrolysis, yielding inositol-1,4,5-trisphosphate and diacylglycerol (DAG) as second messengers to trigger both the increase of intracellular Ca2+ level as well as PKC activation [
It has been, for example, observed that Al3+-induced neural degeneration in rats is greatly enhanced when the animals were fed low doses of fluoride. The presence of fluoride caused more Al3+ to cross the BBB and be deposited in the brain of rats. The formation of AlF4−, according to experimental evidence, in quantities as little as 1 ppm of fluoride contamination of water supplied to rats led to a greater uptake of Al3+ into the brain along with the formation of amyloid deposits like those in Alzheimer's disease.[
Amplification of false message of AlF4− in signaling cascades
The effects of AlF4− are amplified by processes of signal transduction [
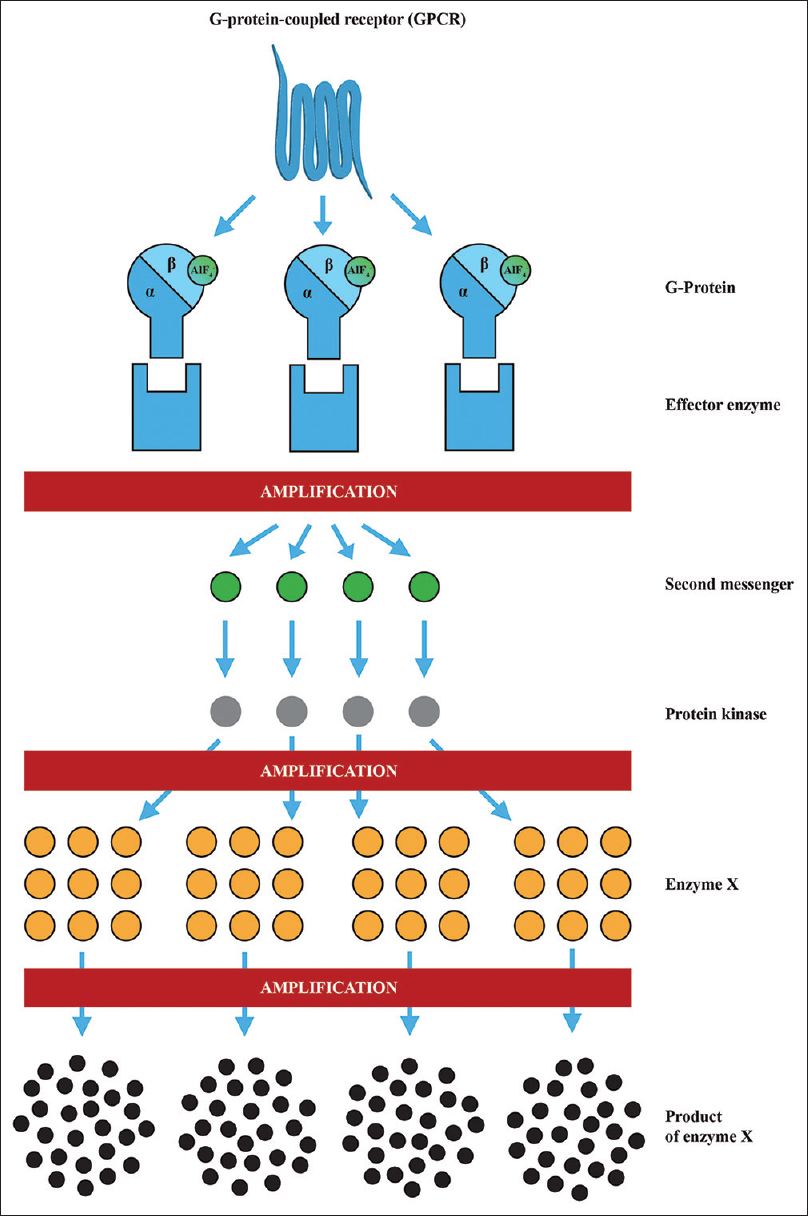
Figure 12
AlF4- acts as the messenger of false information. Its message is greatly amplified during the conversion into the functional response of a cell. Effector enzymes are adenylyl cyclase or phospholipase C, the second messenger molecule could be cAMP, inositol 1,4,5-trisphosphate and diacylglycerol
Basically, by mimicking the mechanism that activates G-protein signaling, aluminofluoride can activate several reactions utilizing G-protein signaling, such as metabotropic glutamate receptors. Protein kinases are essential for GPCRs-mediated signal transduction. AlF4− mimics the transition state of protein phosphorylation. Protein phosphorylation is also important in posttranslational modification of GluRs. The studies of mGluRs show that like many GPCRs they can activate more than a single G protein, can produce various second messengers, and interact with several common protein kinases. The effects of AlF4− might thus result in numerous pathophysiological consequences at several times lower concentrations than either Al3+ or fluoride acting alone. Several common protein kinases, including protein kinase A (PKA), PKC, and mitogen-activated protein kinases (MAPKs), directly interact with mGluR1/5, phosphorylate specific serine or threonine sites, and thereby regulate trafficking, distribution, and function of mGluR1/5 receptors.[
Ji et al., examined alterations in the activity of cAMP-dependent PKA (Protein Kinase A) and PKC (Protein Kinase C) in postmortem brain tissue samples from the frontal, temporal, parietal, and occipital cortices, and the cerebellum of individuals with regressive autism as compared to age-matched control subjects and autistic subjects without clinical history of regression.[
In another trial, Akinrinade et al., treated male adult Wistar rats with low and high doses of fluoride, Al3+ or a combination of both elements for 30 days and compared their effects on brain homogenates.[
In recent years, our understanding of function of G-protein coupled receptors (GPCRs) has changed from a picture of simple signal relays, transmitting only a particular signal to a particular G protein, to versatile machines, capable of various responses to different stimuli and being modulated by various factors. Malfunction of myriads of GPCRs functions is prevalent in human diseases, so that approximately half (estimates vary between 30 and 60%) of marketed drugs target GPCRs.[
The synergistic action of fluoride and micromolar Al3+ can thus evoke a whole network of pathological events. Simultaneously, the heterogeneity of their mutual dynamic interactions can explain the clinically heterogeneous symptoms of ASD and contribute to an understanding of the various responses in any given child to the identical environmental neurotoxins, since we know that each case of autism is unique.
PREVENTION AND AMELIORATION OF AUTISM SPECTRUM DISORDER SYMPTOMS
Recently, Lyra et al. published a review called What do Cochrane systematic reviews say about interventions for autism spectrum disorders?[
HYPOTHESIS FOR THE DEVELOPMENT OF AUTISM SPECTRUM DISORDER FROM AVAILABLE EVIDENCE AND POSSIBLE METHODS OF REVERSAL OF THE SYMPTOMATIC EXPRESSION OF AUTISM SPECTRUM DISORDER AFFECTED NERVOUS SYSTEM SUPPORTED BY OBSERVATIONS FROM THE LITERATURE
Based on our hypothesis of immunoexcitotoxicity we suggest that it is reasonable to conclude that infants and children are exposed to a number of environmental and food-based excitotoxin additives, or conditions that can worsen intrinsic brain inflammation and excitotoxicity. What has emerged is the insight by most investigators that ASD is a complex, multisystem disorder. Disruptions in energy metabolism resulting in mitochondrial dysfunction, impairment in critical regulation of oxidative stress with disturbances of transmethylation/transsulfuration system and decreased GSH level, produce a systemic disorder that affects brain development and function. Basic to this hypothesis is the finding that one sees extensive and prolonged microglial and astrocytic activation and resulting inflammation/excitotoxicity in most cases of ASD. In this review, we show that processes of immunoexcitotoxocity are both triggered and amplified by the environmental toxin fluoride and Al3+.
Reversal of fluoride induced cell injury and fluorosis through the elimination of fluoride and consumption of a diet containing essential nutrients and antioxidants, have been shown by studies in India to be beneficial to brain function, where millions of people suffer with fluorosis.[
Role of nutrition in brain development and the role of Vitamins C, E, and D3, as antioxidants, in reversing the metabolic changes found in fluoride intoxication
Early nutrition has been shown to play a major role in the brain development, mental and immune function. Children and adolescents with poor nutritional status are exposed to alterations of mental and behavioral functions that can be corrected to a certain extent by dietary measures.[
Role of vitamin B6 and vitamin B12, in treating immunoexcitotoxicity
Vitamin B6 can dramatically lower blood and tissue glutamate levels and raise seizure thresholds. High dose of vitamin B6's ability to powerfully inhibit excitotoxicity at least partially explains the often dramatic results reported by Rimland in treating autistic children.[
Role of vitamin D3 in treating autism spectrum disorder
Vitamin D3 deficiency is accounted among the potential environmental risk factors for ASD.[
Saad et al. published a controlled RTC study on 122 ASD children and found that 57% of the patients had vitamin D deficiency, and 30% had vitamin D insufficiency.[
Role of diets in autism spectrum disorder
Many of the diets now being proposed for autistic children emphasize elimination of foods known to be exceedingly high in excitotoxin additives. They are also low in sugar. Jones et al. demonstrated that children respond to glucose challenges with a hypoglycemic response at higher levels of blood glucose than adults.[
Other neutraceuticals that may be of benefit in treating autism spectrum disorder
A number of nutraceuticals have been shown to improve mitochondrial function, including thiamine, riboflavin-5 phosphate, pyridoxal-5 phosphate, ascorbate, acetyl-L-carnitine, pyruvate, malate, CoQ10, curcumin, niacinamide, ketones, DHA, vitamins K, folate and methylcobalamin.[
Taurine may play a crucial role in the protection of antioxidant system and ATPases against Al3+-induced toxicity in brain and blood of rats. Deficiency of taurine in a cat leads to immune activation in the CNS with Purkinje cell loss, microglial activation and astrocytosis, similar to that observed in the ASD brain.[
Many parents of children with ASD report that their child has stool with a sand-like appearance. One of the most common causes comes from a lack of bile acid formation in the liver. Taurine conjugates with bile acids and increases their pool size. Some autism intervention specialist therefore recommend supplementation with taurine.
There is a need for a non-invasive method to both reduce the absorption of Al3+ in the gastrointestinal system and facilitate the excretion of systemic Al3+ in the urine, especially in children, pregnant mothers and women of childbearing age who may become pregnant. Based on the knowledge that silicon is the natural antagonist to Al3+, some researchers have shown that silicon-rich mineral waters can be used to reduce the body burden of Al3+ in individuals with Alzheimer's disease, macrophagic myofasciitis, and chronic fatigue syndrome.[
Pogue and Lukiw[
Mg2+ and zinc also powerfully inhibit excitotoxicity as well as act as co-factors in numerous enzyme systems. Low Mg2+ is associated with dramatic increases in free radical generation as well as GSH depletion. In addition, low Mg2+ enhances NMDA sensitivity, making excitotoxicity more likely even in the presence of physiological levels of extracellular glutamate. High glutamate levels have also been shown to deplete cellular GSH.
Of great interest is the use of selected flavonoids as antioxidants, anti-inflammatories, GluR blockers, and antimicrobials. The flavonoids are more powerful and versatile as antioxidants than are the vitamins.[
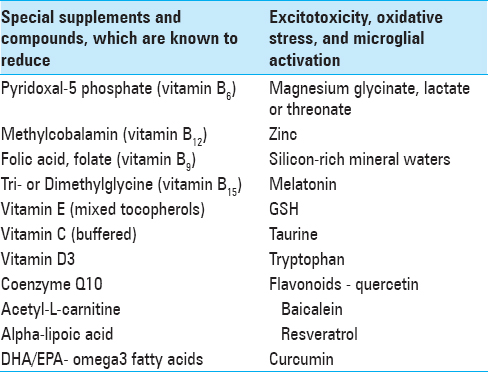
Supplementation with the amino acid tryptophan is considered in the treatment of depression and sleep disorders, mainly due to its relationship with the synthesis of serotonin and melatonin. It is also used in helping to resolve cognitive disorders, anxiety, or neurodegenerative diseases.[
In August 2017, Li with co-workers[
Value of melatonin in the treatment of autism spectrum disorder
Rossignol and Frye evaluated twenty clinical studies, which have reported improvements in sleep parameters with exogenous melatonin supplementation in ASD, including longer sleep duration, less nighttime awakenings and quicker sleep onset.[
CONCLUSIONS
The etiology and pathogeneis of ASD are not well understood. Autism Spectrum Disorders comprise a complex of clinical syndromes found predominantly in infants and younger people consisting of disturbances in cognition and comprehension, communication disorders, epilepsy, and behavior that have an underlying common pathology of neurodegeneration of the cerebellar Purkinje cells and other areas of the brain. Other studies in humans have shown chronic inflammatory changes in the brain, particularly in the cerebellum, and involvement of the microglia in this pathology.
Our review presents evidence suggesting a hypothesis unifying the syndromes in ASD. The clinical as well as pathological findings of the ASD have a set of pathological events with the common denominator being immunoexcitotoxicity leading to neurodegeneration and abnormalities in the connectome, particularly in developing brains in the neonate and young with evidence that explains the clinical presentation of ASD.
Our hypothesis is supported by experimental evidence from animal models and by some clinical testing and pathology studies that give credibility to the concept of immunoexcitotoxicity as the underlying cause of ASD. A great number of conditions can trigger both the inflammatory and the excitotoxic cascade, including sequential vaccination, infections, hypomagnesemia, ROS, RNS and LPP, fluoride and Al3+ as well as a number of other neurotoxic metals and industrial chemicals. Chronic activation of the brain's immune system increases extracellular glutamate levels sufficiently to trigger the excitotoxic cascade, which in conjunction with inflammatory cytokines and prostaglandins, magnifies the damaging effects of both. This mechanism explains most of the features of the ASD, including the behavioral difficulties, language problems, repetitive behaviors, intellectual delay, and episodic dyscontrol of anger. In addition, these mechanisms explain the pathological findings as well, including the changes in the cerebellum, abnormalities in connectivity and the widespread activation of microglia and astrocytes. It also explains why ASD has not disappeared despite the removal of mercury from most childhood vaccines, since excessive immune activation is the initiating and sustaining event in ASD. Evidence is presented that the abundance of fluoride added to the water worldwide and the widespread availability of aluminum particularly to infants and young children through aluminum containing vaccinations, singly or together as aluminofluoride can be potent factors in producing the condition of immunoexcitotoxicity that leads to the pathological changes seen in ASD. The vaccination program should be evaluated to reduce the excessive stimulation of immature immune system and to replace Al3+-adjuvants.
We have also reviewed studies that indicate that fluoride and Al3+, as ubiquitous environmental and food-derived toxins, can exacerbate the pathological and clinical problems of ASD. In synergistic action as AlF4− these elements induce numerous chronic pathophysiological consequences at several times lower concentrations than either Al3+ or fluoride acting alone. AlF4− may evoke several signaling disorders and act as an endocrine disruptor. Moreover, most of the excitotoxic events may enhance the subclinical pathological alterations and/or the genetic susceptibility seen in ASD. The full genetic potential of the child for brain and mental development may be also compromised due to deficiency of micronutrients.
Our immunoexcitotoxic hypothesis opens the door to a number of new modes of prevention and amelioration of ASD. Elimination of various sources of fluoride and Al3+ in early development and consumption of a diet containing essential nutrients and antioxidants have been shown to be beneficial to brain function. As a multifaceted disorder, ASD requires a multifaceted approach, one that should include the protection against excitotoxicity, as well as the protection against microglial activation.
There are several experimental studies that can be constructed to test the immunoexcitotoxic hypothesis, especially as regards vaccines. Such a study would require the use of non-human primates at various stages of development, from intrauterine life to early post-natal development corresponding to human neurodevelopment during vulnerable neurodevelopmental milestones. The study would follow the vaccine schedules used with human newborns and pre-schoolers. One would need to use the exact vaccines used in the human vaccine schedules but of comparable doses based on weight.
To measure microglial activation in response to the vaccines, it would be necessary to use microglial activation PET scanning techniques at various time schedules before and following the vaccines. Newer microglial activation PET scanning techniques are being developed that hold the possibility of differentiating between M1 and M2 microglial activation phenotypes. Long-term follow-up scanning would be necessary to delineate prolonged microglial activation as has been observed in cases of autism. Measure of glutamate levels in the affected areas of the brain should also be conducted, perhaps in a separate set of monkeys to prevent inadvertent activation of local microglia. More extensive studies could be envisioned, such as measures of EAATs, glutaminase, and NADPH oxidase within affected brain areas in response to vaccination.
In addition, studies should be conducted measuring the aluminum, fluoride and aluminofluoride complex concentration in the affected areas of the brain in autopsied cases of autism. This should include the various cell types as well as cellular compartments. This could also be done experimentally in the non-human primate studies using human vaccines described above. Controls could establish the levels of these toxicants to establish the baseline levels in non-vaccinated animals.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Abdulamir HA, Abdul-Rasheed OF, Abdulghani EA. Low oxytocin and melatonin levels and their possible role in the diagnosis and prognosis in Iraqi autistic children. Saudi Med J. 2016. 37: 29-36
2. Abrahám H, Tornóczky T, Kosztolányi G, Seress L. Cell formation in the cortical layers of the developing human cerebellum. Int J Dev Neurosci. 2001. 19: 53-62
3. Adams JB, Audhya T, McDonough-Means S, Rubin RA, Quig D, Geis E. Nutritional and metabolic status of children with autism vs. Neurotypical children, and the association with autism severity. Nutr Metab (Lond). 2011. 8: 34-
4. Adams J, Howsmon DP, Kruger U, Geis E, Gehn E, Fimbres V. Significant association of urinary toxic metals and autism-related symptoms-a nonlinear statistical analysis with cross validation. PLoS One. 2017. 12: e0169526-
5. Adedara IA, Olabiyi BF, Ojuade TD, Idris UF, Onibiyo EM, Farombi EO. Taurine reverses sodium fluoride-mediated increase in inflammation, caspase-3 activity, and oxidative damage along the brain-pituitary-gonadal axis in male rats. Can J Physiol Pharmacol. 2017. 95: 1019-29
6. Akaike A, Tamura Y, Sato Y, Yokota T. Protective effects of a vitamin B12 analog, methylcobalamin, against glutamate cytotoxicity in cultured cortical neurons. Eur J Pharmacol. 1993. 241: 1-6
7. Akinrinade ID, Memudu AE, Ogundele OM, Ajetunmobi OI. Interplay of glia activation and oxidative stress formation in fluoride and aluminium exposure. Pathophysiology. 2015. 22: 39-48
8. Allen G, Courchesne E. Differential effects of developmental cerebellar abnormality on cognitive and motor functions in the cerebellum: An fMRI study of autism. Am J Psychiatry. 2003. 160: 262-73
9. Anitha A, Nakamura K, Thanseem I, Yamada K, Iwayama Y, Toyota T. Brain region-specific altered expression and association of mitochondria-related genes in autism. Mol Autism. 2012. 3: 12-
10. Appel SH, Zhao W, Beers DR, Henkel JS. The microglial motor neuron dialogue in ALS. Acta Myol. 2011. 30: 4-8
11. Aravind A, Dhanya RS, Narayan A, Sam G, Adarsh VJ, Kiran M. Effect of fluoridated water on intelligence in 10-12-year-old school children. J Int Soc Prev Community Dent. 2016. 6: S237-42
12. Aschner M, Du YL, Gannon M, Kimelberg HK. Methylmercury-induced alterations in excitatory amino acid transport in rat primary astrocyte cultures. Brain Res. 1993. 602: 181-6
13. Ashwood P, Van de Water J. A review of autism and the immune response. Clin Dev Immunol. 2004. 11: 165-74
14. Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci. 2001. 21: 6480-91
15. Banala RR, Karnati PR. Vitamin A deficiency: An oxidative stress marker in sodium fluoride (NAF) induced oxidative damage in developing rat brain. Int J Dev Neurosci. 2015. 47: 298-303
16. Bauman PS, Levine SA. The development of children of drug addicts. Int J Addict. 1986. p. 21:849-63
17. Behar TN1, Scott CA, Greene CL, Wen X, Smith SV, Maric D. Glutamate acting at NMDA receptors stimulates embryonic cortical neuronal migration. J Neurosci. 1999. 19: 4449-61
18. Bellebaum C, Daum I. Cerebellar involvement in executive control. Cerebellum. 2007. 6: 184-92
19. Bhatnagar M, Rao P, Sushma J, Bhatnagar R. Neurotoxicity of fluoride: Neurodegeneration in hippocampus of female mice. Indian J Exp Biol. 2002. 40: 546-54
20. Bilbo SD, Schwarz JM. Early-life programming of later-life brain and behavior: A critical role for the immune system. Front Behav Neurosci. 2009. 3: 14-
21. Bilbo SD, Smith SH, Schwarz JM. A lifespan approach to neuroinflammatory and cognitive disorders: A critical role for glia. J Neuroimmune Pharmacol. 2012. 7: 24-41
22. Biou V, Bhattacharyya S, Malenka RC. Endocytosis and recycling of AMPA receptors lacking GluR2/3. Proc Natl Acad Sci U S A. 2008. 105: 1038-43
23. Bishop NJ, Morley R, Day JP, Lucas A. Aluminum neurotoxicity in preterm infants receiving intravenous-feeding solutions. N Engl J Med. 1997. 336: 1557-61
24. Bjorklund G, Saad K, Chirumbolo S, Kern JK, Geier DA, Geier MR. Immune dysfunction and neuroinflammation in autism spectrum disorder. Acta Neurobiol Exp (Wars). 2016. 76: 257-68
25. Blaylock RL. A possible central mechanism in autism spectrum disorders, part 1. Altern Ther Health Med. 2008. 14: 46-53
26. Blaylock RL. A possible central mechanism in autism spectrum disorders, part 2: Immunoexcitotoxicity. Altern Ther Health Med. 2009. 15: 60-7
27. Blaylock RL. A possible central mechanism in autism spectrum disorders, part 3: The role of excitotoxin food additives and the synergistic effects of other environmental toxins. Altern Ther Health Med. 2009. 15: 56-60
28. Blaylock RL. Immunology primer for neurosurgeons and neurologists part 2: Innate brain immunity. Surg Neurol Int. 2013. 4: 118-
29. Blaylock RL. Parkinson's Disease: Microglial/Macrophage-induced immunoexcitotoxicity as a central mechanism of neurodegeneration. Surg Neurol Inter. 2017. 8: 65-
30. Blaylock RL. Phytonutrients and metabolic stimulants as protection against neurodegeneration and excitotoxicity. JAMA. 2000. 2: 30-41
31. Blaylock RL, Maroon J. Immunoexcitotoxicity as a central mechanism in chronic traumatic encephalopathy–a unifying hypothesis. Surg Neurol Inter. 2011. 2: 107-
32. Blaylock RL, Maroon J. Natural plant products and extracts that reduce immunoexcitotoxicity-associated neurodegeneration and promote repair within the central nervous system. Surg Neurol Int. 2012. 3: 19-
33. Blaylock RL, Strunecka A. Immune-glutamatergic dysfunction as a central mechanism of the autism spectrum disorders. Curr Med Chem. 2009. 16: 157-70
34. Bondy SC. Prolonged exposure to low levels of aluminum leads to changes associated with brain aging and neurodegeneration. Toxicology. 2014. 315: 1-7
35. Bourgeois JP, Rakic P. Changes of synaptic density in the primary visual cortex of the macaque monkey from fetal to adult stage. J Neurosci. 1993. 13: 2801-20
36. Bourre JM. Effects of nutrients (in food) on the structure and function of the nervous system: Update on dietary requirements for brain. Part 1: Micronutrients. J Nutr Health Aging. 2006. 10: 377-85
37. Bruttger J, Karram K, Wörtge S, Regen T, Marini F, Hoppmann N. Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity. 2015. 43: 92-106
38. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. 2014. 17: 131-43
39. Butts T, Green MJ, Wingate RJ. Development of the cerebellum: Simple steps to make a ‘little brain'. Development. 2014. 141: 4031-41
40. Cadusseau J, Ragunathan-Thangarajah N, Surenaud M, Hue S, Authier FJ, Gherardi RK. Selective elevation of circulating CCL2/MCP1 levels in patients with longstanding post-vaccinal macrophagic myofasciitis and ASIA. Curr Med Chem. 2014. 21: 511-7
41. Careaga M, Ashwood P. Autism spectrum disorders: From immunity to behavior. Methods Mol Biol. 2012. 934: 219-40
42. Carlsson A. Current problems relating to the pharmacology and toxicology of fluorides. J Swedish Med Assoc. 1978. 14: 1338-92
43. Catania MV, De Socarraz H, Penney JB, Young AB. Metabotropic glutamate receptor heterogeneity in rat brain. Mol Pharmacol. 1994. 45: 626-36
44. Chauhan A, Audhya T, Chauhan V. Brain region-specific glutathione redox imbalance in autism. Neurochem Res. 2012. 37: 1681-9
45. Chauhan A, Gu F, Essa MM, Wegiel J, Kaur K, Brown WT, Chauhan V. Brain region-specific deficit in mitochondrial electron transport chain complexes in children with autism. J Neurochem. 2011. 117: 209-20
46. Chen N, Bao Y, Xue Y, Sun Y, Hu D, Meng S. Meta-analyses of RELN variants in neuropsychiatric disorders. Behav Brain Res. 2017. 332: 110-9
47. Chen R, Zhao LD, Liu H, Li HH, Ren C, Zhang P. Fluoride induces neuroinflammation and alters Wnt signaling pathway in BV2 microglial cells. Inflammation. 2017. 40: 1123-30
48. Chinoy NJ, Tripathi G.editors. Studies on fluoride, aluminium and arsenic toxicity in mammals and amelioration by some antidotes. Modern Trends in Environmental Biology. New Delhi: CBS Publishers; 2002. p. 164-96
49. Chinoy NJ, Sharma AK. Amelioration of fluoride toxicity by vitamin E and D in reproductive function of male mice. Fluoride. 1998. 31: 203-16
50. Choi AL, Sun G, Zhang Y, Grandjean P. Developmental fluoride neurotoxicity: A systematic review and meta-analysis. Environ Health Perspect. 2012. 120: 1362-8
51. Choi AL, Zhang Y, Sun G, Bellinger DC, Wang K, Yang XJ. Association of lifetime exposure to fluoride and cognitive functions in chinese children: A pilot study. Neurotoxicol Teratol. 2015. 47: 96-101
52. Chuchu N, Patel B, Sebastian B, Exley C. The aluminium content of infant formulas remains too high. BMC Pediatr. 2013. 13: 162-
53. Chugani DC, Sundram BS, Behen M, Lee ML, Moore GJ. Evidence of altered energy metabolism in autistic children. Prog Neuropsychopharmacol Biol Psychiatry. 1999. 23: 635-41
54. Cieslinska A, Kostyra E, Chwala B, Moszynska-Dumara M, Fiedorowicz E, Teodorowicz M, Savelkoul HFJ. Vitamin D receptor gene polymorphisms associated with childhood autism. Brain Sci. 2017. 7:
55. Contestabile A. Roles of NMDA receptor activity and nitric oxide production in brain development. Brain Res Brain Res Rev. 2000. 32: 476-509
56. Courchesne E. Brainstem, cerebellar and limbic neuroanatomical abnormalities in autism. Curr Opin Neurobiol. 1997. 7: 269-78
57. Cull-Candy S, Kelly L, Farrant M. Regulation of Ca2+-permeable AMPA receptors: Synaptic plasticity and beyond. Curr Opin Neurobiol. 2006. 16: 288-97
58. Cunningham CL, Martinez-Cerdeno V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci. 2013. 33: 4216-33
59. Damarla SR, Keller TA, Kana RK, Cherkassky VL, Williams DL, Minshew NJ. Cortical underconnectivity coupled with preserved visuospatial cognition in autism: Evidence from an fMRI study of an embedded figures task. Autism Res. 2010. 3: 273-279
60. D'Arcangelo G, Lossi L, Merighi A. Editorial: Reelin-related neurological disorders and animal models. Front Cell Neurosci. 2016. 10: 299-
61. Davenward S, Bentham P, Wright J, Crome P, Job D, Polwart A. Silicon-rich mineral water as a non-invasive test of the ‘aluminum hypothesis’ in Alzheimer's disease. J Alzheimers Dis. 2013. 33: 423-30
62. Dec K, Łukomska A, Maciejewska D, Jakubczyk K, Baranowska-Bosiacka I, Chlubek D. The influence of fluorine on the disturbances of homeostasis in the central nervous system. Biol Trace Elem Res. 2017. 177: 224-34
63. Delhey L, Kilinc EN, Yin L, Slattery J, Tippett M, Wynne R. Bioenergetic variation is related to autism symptomatology. Metab Brain Dis. 2017. 32: 2021-31
64. Delhey LM, Nur Kilinc E, Yin L, Slattery JC, Tippett ML, Rose S. The effect of mitochondrial supplements on mitochondrial activity in children with autism spectrum disorder. J Clin Med. 2017. 6:
65. Dolske MC, Spollen J, McKay S, Lancashire E, Tolbert L. A preliminary trial of ascorbic acid as supplemental therapy for autism. Prog Neuropsychopharmacol Biol Psychiatry. 1993. 17: 765-74
66. Du L. The effect of fluorine on the developing human brain. Zhonghua Bing Li Xue Za Zhi. 1992. 21: 218-
67. Ebisch SJ, Gallese V, Willems RM, Mantini D, Groen WB, Romani GL. Altered intrinsic functional connectivity of anterior and posterior insula regions in high-functioning participants with autism spectrum disorder. Hum Brain Mapp. 2011. 32: 1013-28
68. Edminson E, Ashwood P, Van de Water J. Autoimmunity, autoantibodies, and autism spectrum disorder. Biol Psych. 2017. 81: 383-90
69. Ehlers MD, Fung ET, O'Brien RJ, Huganir RL. Splice variant-specific interaction of the NMDA receptor subunit NR1 with neuronal intermediate filaments. J Neurosci. 1998. 18: 720-30
70. Ekambaram P, Paul V. Modulation of fluoride toxicity in rats by calcium carbonate and by withdrawal of fluoride exposure. Pharmacol Toxicol. 2002. 90: 53-8
71. Endoh T. Characterization of modulatory effects of postsynaptic metabotropic glutamate receptors on calcium currents in rat nucleus tractus solitarius. Brain Res. 2004. 1024: 212-24
72. Last accessed on 2010 Nov 15. https://www.epagov/dwstandardsregulations/fluoride-risk-assessment-and-relative-source-contribution.
73. Estes ML, McAllister AK. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat Rev Neurosci. 2015. 16: 469-86
74. Exley C. Why industry propaganda and political interference cannot disguise the inevitable role played by human exposure to aluminum in neurodegenerative diseases, including Alzheimer's disease. Front Neurol. 2014. 5: 212-
75. Fatemi SH, Aldinger KA, Ashwood P, Bauman ML, Blaha CD, Blatt GJ. Consensus paper: Pathological role of the cerebellum in autism. Cerebellum. 2012. 11: 777-807
76. Fatemi SH, Snow AV, Stary JM, Araghi-Niknam M, Reutiman TJ, Lee S. Reelin signaling is impaired in autism. Biol Psychiatry. 2005. 57: 777-87
77. Fedder KN, Sabo SL. On the role of glutamate in presynaptic development: Possible contributions of presynaptic NMDA receptors. Biomolecules. 2015. 5: 3448-66
78. Ferguson AR, Bolding KA, Huie JR, Hook MA, Santillano DR, Miranda RC. Group I metabotropic glutamate receptors control metaplasticity of spinal cord learning through a protein kinase C-dependent mechanism. J Neurosci. 2008. 28: 11939-49
79. Floden AM, Li S, Combs CK. Beta-amyloid-stimulated microglia induce neuron death via synergistic stimulation of tumor necrosis factor alpha and NMDA receptors. J Neurosci. 2005. 25: 2566-75
80. Ford TC, Nibbs R, Crewther D.P.. Increased glutamate/GABA+ratio in a shared autistic and schizotypal trait phenotype termed Social Disorganisation. Neuroimage Clin. 2017. 16: 125-31
81. Frye RE, Rossignol D.A.. Mitochondrial dysfunction can connect the diverse medical symptoms associated with autism spectrum disorders. Pediatr Res. 2011. 69: 41R-47R
82. Frye RE, Rossignol DA. Treatments for biomedical abnormalities associated with autism spectrum disorder. Front Pediatr. 2014. 2: 66-
83. Frye RE, Wynne R, Rose S, Slattery J, Delhey L, Tippett M. Thyroid dysfunction in children with autism spectrum disorder is associated with folate receptor alpha autoimmune disorder. J Neuroendocrinol. 2017. 29:
84. Furuta A, Martin LJ. Laminar segregation of the cortical plate during corticogenesis is accompanied by changes in glutamate receptor expression. J Neurobiol. 1999. 39: 67-80
85. Furuta A, Rothstein JD, Martin LJ. Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci. 1997. 17: 8363-75
86. Garcimartín A, Merino JJ, Santos-López JA, López-Oliva ME, González MP, Sánchez-Muniz FJ. Silicon as neuroprotector or neurotoxic in the human neuroblastoma SH-SY5Y cell line. Chemosphere. 2015. 135: 217-24
87. Geier DA, Kern JK, Garver CR, Adams JB, Audhya T, Geier MR. A prospective study of transsulfuration biomarkers in autistic disorders. Neurochem Res. 2009. 34: 386-93
88. Gesundheit B, Rosenzweig JP, Naor D, Lerer B, Zachor DA, Procházka V. Immunological and autoimmune considerations of autism spectrum disorders. J Autoimmun. 2013. 44: 1-7
89. Ghanizadeh A. Increased glutamate and homocysteine and decreased glutamine levels in autism: A review and strategies for future studies of amino acids in autism. Dis Markers. 2013. 35: 281-6
90. Gherardi RK, Eidi H, Crépeaux G, Authier FJ, Cadusseau J. Biopersistence and brain translocation of aluminum adjuvants of vaccines. Front Neurol. 2015. 6: 4-
91. Ghiani CA, Beltran-Parrazal L, Sforza DM, Malvar JS, Seksenyan A, Cole R. Genetic program of neuronal differentiation and growth induced by specific activation of NMDA receptors. Neurochem Res. 2007. 32: 363-76
92. Gillberg IC, Gillberg C, Kopp S. Hypothyroidism and autism spectrum disorders. J Child Psychol Psychiatry. 1992. 33: 531-42
93. Gilman AG. G proteins: Transducers of receptor-generated signals. Annu Rev Biochem. 1987. 56: 615-49
94. Ginsberg MR, Rubin RA, Falcone T, Ting AH, Natowicz MR. Brain transcriptional and epigenetic associations with autism. PLoS One. 2012. 7: e44736-
95. Goines PE, Ashwood P. Cytokine dysregulation in autism spectrum disorders (ASD): Possible role of the environment. Neurotoxicol Teratol. 2013. 36: 67-81
96. Gomez-Gonzalez B, Escobar A. Prenatal stress alters microglial development and distribution in postnatal rat brain. Acta Neuropathol. 2010. 119: 303-15
97. Grandjean P, Landrigan PJ. Neurobehavioural effects of developmental toxicity. Lancet Neurol. 2014. 13: 330-8
98. Gregg C, Weiss S. CNTF/LIF/gp130 receptor complex signaling maintains a VZ precursor differentiation gradient in the developing ventral forebrain. Development. 2005. 132: 565-78
99. Guevara-Campos J, González-Guevara L, Cauli O. Autism and intellectual disability associated with mitochondrial disease and hyperlactacidemia. Int J Mol Sci. 2015. 16: 3870-
100. Guna-Sherlin DM, Verma RJ. Vitamin D ameliorates fluoride-induced embryotoxicity in pregnant rats. Neurotoxicol Teratol. 2001. 23: 197-201
101. Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat Commun. 2014. 5: 5748-
102. Hamada S, Ogawa I, Yamasaki M, Kiyama Y, Kassai H, Watabe AM. The glutamate receptor GluN2 subunit regulates synaptic trafficking of AMPA receptors in the neonatal mouse brain. Eur J Neurosci. 2014. 40: 3136-46
103. Hamm HE. The many faces of G protein signaling. J Biol Chem. 1998. 273: 669-72
104. Hasel P, McKay S, Qiu J. Hardingham, G.E Selective dendritic susceptibility to bioenergetic, excitotoxic and redox perturbations in cortical neurons. Biochim Biophys Acta. 2015. 1853: 2066-76
105. Hashimoto T, Aihara R, Tayama M, Miyazaki M, Shirakawa Y, Kuroda Y. Reduced thyroid-stimulating hormone response to thyrotropin-releasing hormone in autistic boys. Dev Med Child Neurol. 1991. 33: 313-9
106. Herrmann K. Differential distribution of AMPA receptors and glutamate during pre- and postnatal development in the visual cortex of ferrets. J Comp Neurol. 1996. p. 375:1-17
107. Hoshiko S, Grether JK, Windham GC, Smith D, Fessel K. Are thyroid hormone concentrations at birth associated with subsequent autism diagnosis?. Autism Res. 2011. 4: 456-63
108. Hsiao EY. Immune dysregulation in autism spectrum disorder. Int Rev Neurobiol. 2013. 113: 269-302
109. Huttenlocher PR. Morphometric study of human cerebral cortex development. Neuropsychologia. 1990. 28: 517-27
110. Huttenlocher PR, Dabholkar AS. Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol. 1997. 387: 167-78
111. Ikuta T, Shafritz KM, Bregman J, Peters BD, Gruner P, Malhotra AK. Abnormal cingulum bundle development in autism: A probabilistic tractography study. Psychiatry Res. 2014. 221: 63-8
112. Imamoto K, Leblond CP. Radioautographic investigation of gliogenesis in the corpus callosum of young rats Ii Origin of microglial cells. J Comp Neurol. 1978. 180: 139-63
113. . C. A genomewide screen for autism: Strong evidence for linkage to chromosomes 2q, 7q, and 16p. Am J Hum Genet. 2001. 69: 570-81
114. James SJ, Cutler P, Melnyk S, Jernigan S, Janak L, Gaylor DW. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr. 2004. 80: 1611-7
115. James SJ, Melnyk S, Fuchs G, Reid T, Jernigan S, Pavliv O. Efficacy of methylcobalamin and folinic acid treatment on glutathione redox status in children with autism. Am J Clin Nutr. 2009. 89: 425-30
116. Ji K, Akgul G, Wollmuth LP, Tsirka SE. Microglia actively regulate the number of functional synapses. PLoS One. 2013. 8: e56293-
117. Ji L, Chauhan A, Chauhan V. Reduced activity of protein kinase C in the frontal cortex of subjects with regressive autism: Relationship with developmental abnormalities. Int J Biol Sci. 2012. 8: 1075-84
118. Ji L, Chauhan V, Flory MJ, Chauhan A. Brain region-specific decrease in the activity and expression of protein kinase A in the frontal cortex of regressive autism. PLoS One. 2011. 6: e23751-
119. Jones TW, Borg WP, Boulware SD, McCarthy G, Sherwin RS, Tamborlane WV. Enhanced adrenomedullary response and increased susceptibility to neuroglycopenia: Mechanisms underlying the adverse effects of sugar ingestion in healthy children. J Pediatr. 1995. 126: 171-7
120. Kaluzna-Czaplinska J, Jozwik-Pruska J, Chirumbolo S, Bjorklund G. Tryptophan status in autism spectrum disorder and the influence of supplementation on its level. Metab Brain Dis. 2017. 32: 1585-93
121. Kater SB, Mills LR. Regulation of growth cone behavior by calcium. J Neurosci. 1991. 11: 891-9
122. Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996. 274: 1133-8
123. Kemper TL, Bauman ML. Neuropathology of infantile autism. Mol Psychiatry. 2002. 7: S12-3
124. Kerley CP, Power C, Gallagher L, Coghlan D. Lack of effect of vitamin D3 supplementation in autism: A 20-week, placebo-controlled rct. Arch Dis Child. 2017. 102: 1030-6
125. Khan SA, Singh RK, Navit S, Chadha D, Johri N, Navit P. Relationship between dental fluorosis and intelligence quotient of school going children in and around lucknow district: A cross-sectional study. J Clin Diagn Res. 2015. 9: ZC10-5
126. Khan Z, Combadière C, Authier FJ, Itier V, Lux F, Exley C. Slow CCL2-dependent translocation of biopersistent particles from muscle to brain. BMC Med. 2013. 11: 99-
127. Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG. Microglia emerge from erythromyeloid precursors via pu.1- and irf8-dependent pathways. Nat Neurosci. 2013. 16: 273-80
128. Kim H, Lee Y, Park JY, Kim JE, Kim TK, Choi J. Loss of adenylyl cyclase type-5 in the dorsal striatum produces autistic-like behaviors. Mol Neurobiol. 2017. 54: 7994-8008
129. Kočovská E, Gaughran F, Krivoy A, Meier UC. Vitamin-D deficiency as a potential environmental risk factor in multiple sclerosis, schizophrenia, and autism. Front Psychiatry. 2017. 8: 47-
130. Komuro H, Rakic P. Modulation of neuronal migration by NMDA receptors. Science. 1993. 260: 95-7
131. Konur S, Ghosh A. Calcium signaling and the control of dendritic development. Neuron. 2005. 46: 401-5
132. Kostovic I, Judas M. Correlation between the sequential ingrowth of afferents and transient patterns of cortical lamination in preterm infants. Anat Rec. 2002. 267: 1-6
133. Krakowiak P, Goines PE, Tancredi DJ, Ashwood P, Hansen RL, Hertz-Picciotto I. Neonatal cytokine profiles associated with autism spectrum disorder. Biol Psychiatry. 2017. 81: 442-51
134. Kugler P, Schleyer V. Developmental expression of glutamate transporters and glutamate dehydrogenase in astrocytes of the postnatal rat hippocampus. Hippocampus. 2004. 14: 975-85
135. Kumada T, Komuro H. Completion of neuronal migration regulated by loss of Ca(2+) transients. Proc Natl Acad Sci USA. 2004. 101: 8479-84
136. Kumar V, Bal A, Gill KD. Impairment of mitochondrial energy metabolism in different regions of rat brain following chronic exposure to aluminium. Brain Res. 2008. 1232: 94-103
137. Kumar V, Gill KD. Oxidative stress and mitochondrial dysfunction in aluminium neurotoxicity and its amelioration: A review. Neurotoxicology. 2014. 41: 154-66
138. Lammert DB, Howell BW. RELN mutations in autism spectrum disorder. Front Cell Neurosci. 2016. 10: 84-
139. Lee AS, Azmitia EC, Whitaker-Azmitia PM. Developmental microglial priming in postmortem autism spectrum disorder temporal cortex. Brain Behav Immun. 2017. 62: 193-202
140. Leonoudakis D, Zhao P, Beattie EC. Rapid tumor necrosis factor alpha-induced exocytosis of glutamate receptor 2-lacking AMPA receptors to extrasynaptic plasma membrane potentiates excitotoxicity. J Neurosci. 2008. 28: 2119-30
141. Leung CCY, Wong YH. Role of G protein-coupled receptors in the regulation of structural plasticity and cognitive function. Molecules. 2017. 22:
142. Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. 2013. 18: 522-55
143. Lewitus GM, Pribiag H, Duseja R, St-Hilaire M, Stellwagen D. An adaptive role of TNF-alpha in the regulation of striatal synapses. J Neurosci. 2014. 34: 6146-55
144. Li X, Chauhan A, Sheikh AM, Patil S, Chauhan V, Li XM. Elevated immune response in the brain of autistic patients. J Neuroimmunol. 2009. 207: 111-6
145. Li YJ, Ou JJ, Li YM, Xiang DX. Dietary supplement for core symptoms of autism spectrum disorder: Where are we now and where should we go?. Front Psychiatry. 2017. 8: 155-
146. Liu R, Liu IY, Bi X, Thompson RF, Doctrow SR, Malfroy B. Reversal of age related learning deficits and brain oxidative stress in mice with superoxide dismutase/catalase mimetics. Proc Natl Acad. Sci USA. 2003. 128: 15103-
147. Lombard J. Autism: A mitochondrial disorder?. Med Hypotheses. 1998. 50: 497-500
148. López-Bendito G, Shigemoto R, Fairén A, Luján R. Differential distribution of group I metabotropic glutamate receptors during rat cortical development. Cereb Cortex. 2002. 12: 625-38
149. LoTurco JJ, Blanton MG, Kriegstein AR. Initial expression and endogenous activation of NMDA channels in early neocortical development. J Neurosci. 1991. 11: 792-9
150. Lucas DR, Newhouse JP. The toxic effect of sodium l-glutamate on the inner layers of the retina. AMA Arch Ophthalmol. 1957. 58: 193-201
151. Luo L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci. 2000. p. 1:173-80
152. Luke J. Fluoride deposition in the aged human pineal gland. Caries Res. 2001. 35: 125-8
153. Luke J.editors. The effect of fluoride on the physiology of the pineal gland. University of Surrey, Guildford: 1997. p.
154. Lyra L, Rizzo LE, Sunahara CS, Pachito DV, Latorraca COC, Martimbianco ALC. What do Cochrane systematic reviews say about interventions for autism spectrum disorders?. Sao Paulo Med J. 2017. 135: 192-201
155. Ma L, Li XW, Zhang SJ, Yang F, Zhu GM, Yuan XB. Interleukin-1 beta guides the migration of cortical neurons. J Neuroinflammation. 2014. 11: 114-
156. Ma NS, Thompson C, Weston S. Brief report: Scurvy as a manifestation of food selectivity in children with autism. J Autism Dev Disord. 2016. 46: 1464-70
157. Ma XC, Gottschall PE, Chen LT, Wiranowska M, Phelps CP. Role and mechanisms of interleukin-1 in the modulation of neurotoxicity. Neuroimmunomodulation. 2002. 10: 199-207
158. Malatesta P, Götz M, Barsacchi G, Price J, Zoncu R, Cremisi F. PC3 overexpression affects the pattern of cell division of rat cortical precursors. Mech Dev. 2000. 90: 17-28
159. Mandic-Maravic V, Pljesa-Ercegovac M, Mitkovic-Voncina M, Savic-Radojevic A, Lecic-Tosevski D, Simic T. Impaired redox control in autism spectrum disorders: Could it be the x in gxe?. Curr Psychiatry Rep. 2017. 19: 52-
160. Marín-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. Microglia promote the death of developing Purkinje cells. Neuron. 2004. 41: 535-47
161. Martin BR. Ternary complexes of Al3+and F- with a third ligand. Coordination Chem Rev. 1996. 149: 23-32
162. Martínez-Galán JR, López-Bendito G, Luján R, Shigemoto R, Fairén A, Valdeolmillos M. Cajal-Retzius cells in early postnatal mouse cortex selectively express functional metabotropic glutamate receptors. Eur J Neurosci. 2001. 13: 1147-54
163. Mattson MP, Kater SB. Excitatory and inhibitory neurotransmitters in the generation and degeneration of hippocampal neuroarchitecture. Brain Res. 1989. 478: 337-48
164. Mead J, Ashwood P. Evidence supporting an altered immune response in ASD. Immunol Lett. 2015. 163: 49-55
165. Mehdi S, Jarvi ET, Koehl JR, McCarthy JR, Bey P. The mechanism of inhibition of S-adenosyl-L-homocysteine hydrolase by fluorine-containing adenosine analogs. J Enzyme Inhib. 1990. 4: 1-13
166. Mehler MF, Marmur R, Gross R, Mabie PC, Zang Z, Papavasiliou A. Cytokines regulate the cellular phenotype of developing neural lineage species. Int J Dev Neurosci. 1995. 13: 213-40
167. Meldrum BS.editors. Glutamate as a neurotransmitter in the brain: Review of physiology and pathology. J Nutr. 2000. 130: 1007S-1015S
168. Miranda-Contreras L, Benítez-Diaz PR, Mendoza-Briceño RV, Delgado-Saez MC, Palacios-Prü EL. Levels of amino acid neurotransmitters during mouse cerebellar neurogenesis and in histotypic cerebellar cultures. Dev Neurosci. 1999. 21: 147-58
169. Mold M, Shardlow E, Exley C. Insight into the cellular fate and toxicity of aluminium adjuvants used in clinically approved human vaccinations. Sci Rep. 2016. 6: 31578-
170. Monier A, Adle-Biassette H, Delezoide AL, Evrard P, Gressens P, Verney C. Entry and distribution of microglial cells in human embryonic and fetal cerebral cortex. J Neuropathol Exp Neurol. 2007. 66: 372-82
171. Morris G, Puri BK, Frye RE. The putative role of environmental aluminium in the development of chronic neuropathology in adults and children. How strong is the evidence and what could be the mechanisms involved?. Metab Brain Dis. 2017. 32: 1335-55
172. Mukandala G, Tynan R, Lanigan S, O'Connor JJ. The effects of hypoxia and inflammation on synaptic signaling in the CNS. Brain Sci. 2016. 6:
173. Nagarajappa R, Pujara P, Sharda AJ, Asawa K, Tak M, Aapaliya P. Comparative assessment of intelligence quotient among children living in high and low fluoride areas of Kutch, India-a pilot study. Iran J Public Health. 2013. 42: 813-8
174. Nakanishi S, Nakajima Y, Masu M, Ueda Y, Nakahara K, Watanabe D. Glutamate receptors: Brain function and signal transduction. Brain Res Brain Res Rev. 1998. 26: 230-5
175. Navascues J, Calvente R, Marin-Teva JL, Cuadros MA. Entry, dispersion and differentiation of microglia in the developing central nervous system. An Acad Bras Cienc. 2000. 72: 91-102
176. Neumann H, Schweigreiter R, Yamashita T, Rosenkranz K, Wekerle H, Barde YA. Tumor necrosis factor inhibits neurite outgrowth and branching of hippocampal neurons by a rho-dependent mechanism. J Neurosci. 2002. 22: 854-62
177. Nevison CD. A comparison of temporal trends in United States autism prevalence to trends in suspected environmental factors. Environ Health. 2014. 13: 73-
178. Nir I, Meir D, Zilber N, Knobler H, Hadjez J, Lerner Y. Brief report: Circadian melatonin, thyroid-stimulating hormone, prolactin, and cortisol levels in serum of young adults with autism. J Autism Dev Disord. 1995. 25: 641-54
179. Ng M, de Montigny JG, Ofner M, Do MT. Environmental factors associated with autism spectrum disorder: A scoping review for the years 2003-2013. Health Promot Chronic Dis Prev Can. 2017. 37: 1-23
180. Noda M. Dysfunction of glutamate receptors in microglia may cause neurodegeneration. Curr Alzheimer Res. 2016. 13: 381-6
181. Norman AW. A vitamin D nutritional cornucopia: New insights concerning the serum 25-hydroxyvitamin D status of the US population. Am J Clin Nutr. 2008. 88: 1455-6
182. Oldreive CE, Doherty GH. Effects of tumour necrosis factor-alpha on developing cerebellar granule and Purkinje neurons in vitro. J Mol Neurosci. 2010. 42: 44-52
183. Olney JW. Glutamate-induced retinal degeneration in neonatal mice. Electron microscopy of the acutely evolving lesion. J Neuropathol Exp Neurol. 1969. 28: 455-74
184. Pagan C, Delorme R, Callebert J, Goubran-Botros H, Amsellem F, Drouot X. The serotonin-n-acetylserotonin-melatonin pathway as a biomarker for autism spectrum disorders. Transl Psychiatry. 2014. 4: e479-
185. Pagan C, Goubran-Botros H, Delorme R, Benabou M, Lemière N, Murray K. Disruption of melatonin synthesis is associated with impaired 14-3-3 and mir-451 levels in patients with autism spectrum disorders. Sci Rep. 2017. 7: 2096-
186. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011. 333: 1456-8
187. Pardo CA, Vargas DL, Zimmerman AW. Immunity, neuroglia and neuroinflammation in autism. Int Rev Psychiatry. 2005. 17: 485-95
188. Park E, Cohen I, Gonzalez M, Castellano MR, Flory M, Jenkins EC. Is taurine a biomarker in autistic spectrum disorder?. Adv Exp Med Biol. 2017. 975: 3-16
189. Pelvig DP, Pakkenberg H, Stark AK, Pakkenberg B. Neocortical glial cell numbers in human brains. Neurobiol Aging. 2008. 29: 1754-62
190. Perez-Pouchoulen M, VanRyzin JW, McCarthy MM. Morphological and phagocytic profile of microglia in the developing rat cerebellum. eNeuro. 2015. 2:
191. Petroff OA. GABA and glutamate in the human brain. Neuroscientist. 2002. 8: 562-73
192. Planerova A, Philip S, Elad S. Gingival bleeding in a patient with autism spectrum disorder: A key finding leading to a diagnosis of scurvy. Quintessence Int. 2017. 48: 407-11
193. Pogue AI, Lukiw WJ. Natural and synthetic neurotoxins in our environment: From Alzheimer's disease (AD) to autism spectrum disorder (ASD). J Alzheimers Dis Parkinsonism. 2016. 6:
194. Pogue AI, Lukiw WJ. The mobilization of aluminum into the biosphere. Front Neurol. 2014. 5: 262-
195. Qian X1, Shen Q, Goderie SK, He W, Capela A, Davis AA. Timing of CNS cell generation: A programmed sequence of neuron and glial cell production from isolated murine cortical stem cells. Neuron. 2000. 28: 69-80
196. Qiao M, Liu P, Ren X, Feng T, Zhang Z. Potential protection of taurine on antioxidant system and ATPase in brain and blood of rats exposed to aluminum. Biotechnol Lett. 2015. 37: 1579-84
197. Rall TW, Sutherland EW. Formation of a cyclic adenine ribonucleotide by tissue particles. J Biol Chem. 1958. 232: 1065-76
198. Rash BG, Ackman JB, Rakic P. Bidirectional radial Ca(2+) activity regulates neurogenesis and migration during early cortical column formation. Sci Adv. 2016. 2: e1501733-
199. Redhead K, Quinlan G, Das R, Gutteridge J. Aluminium-adjuvanted vaccines transiently increase aluminium levels in murine brain tissue. Pharmacol Toxicol. 1992. 70: 278-80
200. Rezaie P, Male D. Colonisation of the developing human brain and spinal cord by microglia: A review. Microscopy Res Tech. 1999. 45: 359-82
201. Rimland B. High dose vitamin B6 and magnesium in treating autism: Response to study by Findling et al . J Autism Dev Disord. 1998. 28: 581-2
202. Ritter LM, Unis AS, Meador-Woodruff JH. Ontogeny of ionotropic glutamate receptor expression in human fetal brain. Brain Res Dev Brain Res. 2001. 127: 123-33
203. Rocha-Amador D, Navarro ME, Carrizales L, Morales R, Calderon J. Decreased intelligence in children and exposure to fluoride and arsenic in drinking water. Cad Saude Publica. 2007. 23: S579-587
204. Rodbell M. Nobel lecture. Signal transduction: Evolution of an idea. Biosci Rep. 1995. 15: 117-33
205. Rose S1, Melnyk S, Pavliv O, Bai S, Nick TG, Frye RE. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl Psychiatry. 2012. 2: e134-
206. Rossignol DA, Frye RE. Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front Physiol. 2014. 5: 150-
207. Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol Psychiatry. 2012. 17: 290-314
208. Saad K, Abdel-Rahman AA, Elserogy YM, Al-Atram AA, El-Houfey AA, Othman HA. Effects of microglia on neurogenesis. Glia. 2015. 63: 1394-405
209. Sabo SL, McAllister AK. Mobility and cycling of synaptic protein-containing vesicles in axonal growth cone filopodia. Nat Neurosci. 2003. 6: 1264-9
210. Sato K. Effects of microglia on neurogenesis. Glia. 2015. 63: 1394-405
211. Savory J, Herman MM, Ghribi O. Supplementation of the diet with silicic acid to reduce body burden of aluminum: A miracle cure or useless treatment for Alzheimer's disease?. J Alzheimers Dis. 2006. 10: 25-7
212. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012. 74: 691-705
213. Schmahmann JD, Sherman JC. The cerebellar cognitive affective syndrome. Brain. 1998. 121: 561-79
214. Schwarz JM, Bilbo SD. Sex, glia, and development: Interactions in health and disease. Horm Behav. 2012. 62: 243-53
215. Schwarz JM, Sholar PW, Bilbo SD. Sex differences in microglial colonization of the developing rat brain. J Neurochem. 2012. 120: 948-63
216. Seraj B, Shahrabi M, Shadfar M, Ahmadi R, Fallahzadeh M, Eslamlu HF. Effect of high water fluoride concentration on the intellectual development of children in Makoo/Iran. J Dent (Tehran). 2012. 9: 221-9
217. Sharma DR, Wani WY, Sunkaria A, Kandimalla RJ, Sharma RK, Verma D. Quercetin attenuates neuronal death against aluminum-induced neurodegeneration in the rat hippocampus. Neuroscience. 2016. 324: 163-76
218. Sharma P, Mishra KP.editors. Aluminum-induced maternal and developmental toxicity and oxidative stress in rat brain; Response to combined administration of tiron and glutathione. Reprod Toxicol. 2006. p. 313-21
219. Shaw CA, Petrik MS. Aluminum hydroxide injections lead to motor deficits and motor neuron degeneration. J Inorg Biochem. 2009. 103: 1555-62
220. Shaw CA, Seneff S, Kette SD, Tomljenovic L, Oller JW, Davidson RM. Aluminum-induced entropy in biological systems: Implications for neurological disease. J Toxicol. 2014. 2014:
221. Shaw WJ, Campbell AA, Paine ML, Snead ML. The COOH terminus of the amelogenin, LRAP, is oriented next to the hydroxyapatite surface. J Biol Chem. 2004. 279: 40263-6
222. Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci. 1997. 17: 7503-22
223. Shimmura C, Suda S, Tsuchiya KJ, Hashimoto K, Ohno K, Matsuzaki H. Alteration of plasma glutamate and glutamine levels in children with high-functioning autism. PLoS One. 2011. 6: e25340-
224. Shinohe A, Hashimoto K, Nakamura K, Tsujii M, Iwata Y, Tsuchiya KJ. Increased serum levels of glutamate in adult patients with autism. Prog Neuropsychopharmacol Biol Psychiatry. 2006. 30: 1472-7
225. Shivarajashankara YM, Shivarajashankara AR, Bhat PG, Rao SH. Brain lipid peroxidation and antioxidant systems of young rats in chronic fluoride intoxication. Fluoride. 2002. 35: 197-203
226. Shivarajashankara YM, Shivashankara AR, Bhat PG, Rao SH. Lipid peroxidation and antioxidant systems in the blood of young rats subjected to chronic fluoride toxicity. Indian J Exp Biol. 2003. 41: 857-60
227. Shivarajashankara YM, Shivarajashankara AR, Gopalakrishna Bhat P, Muddanna Rao S, Hanumanth Rao S. Histological changes in the brain of young fluoride-intoxicated rats. Fluoride. 2002. 35: 12-21
228. Shoenfeld Y, Agmon-Levin N. 'Asia'-autoimmune/inflammatory syndrome induced by adjuvants. J Autoimmun. 2011. 36: 4-8
229. Shuhua X, Ziyou L, Ling Y, Fei W, Sun G. A role of fluoride on free radical generation and oxidative stress in BV-2 microglia cells. Mediators Inflamm. 2012. 2012: 102954-
230. Singer HS, Morris CM, Gause CD, Gillin PK, Crawford S, Zimmerman AW. Antibodies against fetal brain in sera of mothers with autistic children. J Neuroimmunol. 2008. 194: 165-72
231. Singh N, Verma KG, Verma P, Sidhu GK, Sachdeva S. A comparative study of fluoride ingestion levels, serum thyroid hormone & TSH level derangements, dental fluorosis status among school children from endemic and non-endemic fluorosis areas. Springerplus. 2014. 3: 7-
232. Singh S, Yazdani U, Gadad B, Zaman S, Hynan LS, Roatch N. Serum thyroid-stimulating hormone and interleukin-8 levels in boys with autism spectrum disorder. J Neuroinflammation. 2017. 14: 113-
233. Smith AL, Thompson ID. Spatiotemporal patterning of glutamate receptors in developing ferret striate cortex. Eur J Neurosci. 1999. 11: 923-34
234. Squarzoni P, Oller G, Hoeffel G, Pont-Lezica L, Rostaing P, Low D. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 2014. 8: 1271-9
235. Stephan AH, Barres BA, Stevens B. The complement system: An unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012. 35: 369-89
236. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N. The classical complement cascade mediates CNS synapse elimination. Cell. 2007. 131: 1164-78
237. Strunecka A, Blaylock RL, Hyman M, Paclt I, Strunecka A.editors. Cellular and molecular biology of autism spectrum disorders. Bentham Science Pub; 2010. p.
238. Strunecka A, Blaylock RL, Patocka J. Aluminofluoride complexes: Phosphate analogues and a hidden hazard for living organisms. Curr Inorg Chem. 2012. 2: 8-18
239. Strunecka A, Blaylock R, Strunecky O. Fluoride, aluminum, and aluminofluoride complexes in pathogenesis of the autism spectrum disorders: A possible role of immunoexcitotoxicity. J Appl Biomed. 2016. 14: 171-6
240. Strunecka A, Patocka J, Blaylock RL, Chinoy N. Fluoride interactions: From molecules to disease. Curr Sig Transd Ther. 2007. 2: 190-213
241. Strunecká A, Patočka J, Atwood D, Roesky C.editors. Group 13 Chemistry II. Series Structure and Bonding 104. Springer-Verlag; 2003. p. 139-181
242. Strunecka A, Patocka J. Reassessment of the role of aluminum in the development of Alzheimer's disease. Cesk Fysiol. 1999. 48: 9-15
243. Strunecka A, Strunecky O, Patocka J. Fluoride plus aluminum: Useful tools in laboratory investigations, but messengers of false information. Physiol Res. 2002. 51: 557-64
244. Struys-Ponsar C, Guillard O, van den Bosch de Aguilar P. Effects of aluminum exposure on glutamate metabolism: A possible explanation for its toxicity. Exp Neurol. 2000. 163: 157-64
245. Susheela AK. Fluorosis management programme in India. Curr Sci. 1999. 70: 298-304
246. Susheela AK, Bhatnagar M, Vig K, Mondal NK. Excess fluoride ingestion and thyroid hormone derangements in children living in Delhi, India. Fluoride. 2005. 38: 98-108
247. Suzuki K, Sugihara G, Ouchi Y, Nakamura K, Futatsubashi M, Takebayashi K. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry. 2013. 70: 49-58
248. Swinnen N, Smolders S, Avila A, Notelaers K, Paesen R, Ameloot M. Complex invasion pattern of the cerebral cortex by microglial cells during development of the mouse embryo. Glia. 2013. 61: 150-63
249. Tanaka M, Senda T, Hirashima N. Expression of the GluA2 subunit of glutamate receptors is required for the normal dendritic differentiation of cerebellar Purkinje cells. Neurosci Lett. 2017. 657: 22-6
250. Tang G, Gutierrez Rios P, Kuo SH, Akman HO, Rosoklija G, Tanji K. Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiol Dis. 2013. 54: 349-61
251. Tang QQ, Du J, Ma HH, Jiang SJ, Zhou XJ. Fluoride and children's intelligence: A meta-analysis. Biol Trace Elem Res. 2008. 126: 115-20
252. Tárnok K, Czöndör K, Jelitai M, Czirók A, Schlett K. NMDA receptor NR2B subunit over-expression increases cerebellar granule cell migratory activity. J Neurochem. 2008. 104: 818-29
253. Theoharides TC, Athanassiou M, Panagiotidou S, Doyle R. Dysregulated brain immunity and neurotrophin signaling in Rett syndrome and autism spectrum disorders. J Neuroimmunol. 2015. 279: 33-8
254. Tiemeier H, Lenroot RK, Greenstein DK, Tran L, Pierson R, Giedd JN. Cerebellum development during childhood and adolescence: A longitudinal morphometric MRI study. Neuroimage. 2010. 49: 63-70
255. Tissir F, Goffinet AM. Reelin and brain development. Nat Rev Neurosci. 2003. 4: 496-505
256. Tomljenovic L, Shaw CA. Do aluminum vaccine adjuvants contribute to the rising prevalence of autism?. J Inorg Biochem. 2011. 105: 1489-99
257. Tomljenovic L, Shaw CA. Mechanisms of aluminum adjuvant toxicity and autoimmunity in pediatric populations. Lupus. 2012. 21: 223-30
258. Tong CK, Vidyadaran S. Role of microglia in embryonic neurogenesis. Exp Biol Med. 2016. 241: 1669-75
259. Tonhajzerova I, Ondrejka I, Mestanik M, Mikolka P, Hrtanek I, Mestanikova A. Inflammatory activity in autism spectrum disorder. Adv Exp Med Biol. 2015. 861: 93-8
260. Tordjman S, Anderson GM, Pichard N, Charbuy H, Touitou Y. Nocturnal excretion of 6-sulphatoxymelatonin in children and adolescents with autistic disorder. Biol Psychiatry. 2005. 57: 134-8
261. Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol Rev. 2010. 62: 405-96
262. Tremblay ME, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010. 8: e1000527-
263. Vandenberg RJ, Ryan RM. Mechanisms of glutamate transport. Physiol Rev. 2013. 93: 1621-57
264. Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005. 57: 67-81
265. Varner JA, Jensen KF, Horvath W, Isaacson RL. Chronic administration of aluminum-fluoride or sodium- fluoride to rats in drinking water alterations in neuronal and cerebrovascular integrity. Brain Res. 1998. 784: 284-98
266. Veatch OJ, Goldman SE, Adkins KW, Malow BA. Melatonin in children with autism spectrum disorders: How does the evidence fit together?. J Nat Sci. 2015. 1: e125-
267. Verma RJ, Sherlin DM. Vitamin C ameliorates fluoride-induced embryotoxicity in pregnant rats. Hum Exp Toxicol. 2001. 20: 619-23
268. Viezeliene D, Beekhof P, Gremmer E, Rodovicius H, Sadauskiene I, Jansen E. Selective induction of IL-6 by aluminum-induced oxidative stress can be prevented by selenium. J Trace Elem Med Biol. 2013. 27: 226-9
269. Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the src family of kinases. J Neurosci. 2003. 23: 8692-700
270. Walton JR. Brain lesions comprised of aluminum-rich cells that lack microtubules may be associated with the cognitive deficit of Alzheimer's disease. Neurotoxicology. 2009. 30: 1059-69
271. Walton JR. Functional impairment in aged rats chronically exposed to human range dietary aluminum equivalents. Neurotoxicology. 2009. 30: 182-93
272. Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev. 2005. 85: 1159-204
273. Willard SS, Koochekpour S. Glutamate, glutamate receptors, and downstream signaling pathways. Int J Biol Sci. 2013. 9: 948-59
274. Wills S, Cabanlit M, Bennett J, Ashwood P, Amaral DG, Van de Water J. Detection of autoantibodies to neural cells of the cerebellum in the plasma of subjects with autism spectrum disorders. Brain Behav Immun. 2009. 23: 64-74
275. Wong HH, Smith RG. Patterns of complementary and alternative medical therapy use in children diagnosed with autism spectrum disorders. J Autism Dev Disord. 2006. 36: 901-9
276. Yamada H, Yatsushiro S, Ishio S, Hayashi M, Nishi T, Yamamoto A. Metabotropic glutamate receptors negatively regulate melatonin synthesis in rat pinealocytes. J Neurosci. 1998. 18: 2056-62
277. Yan L, Liu S, Wang C, Wang F, Song Y, Yan N. JNK and NADPH oxidase involved in fluoride-induced oxidative stress in BV-2 microglia cells. Mediators Inflamm. 2013. 2013: 895975-
278. Yan N, Liu Y, Liu S, Cao S, Wang F, Wang Z. Fluoride-induced neuron apoptosis and expressions of inflammatory factors by activating microglia in rat brain. Mol Neurobiol. 2016. 53: 4449-60
279. Yawata I, Takeuchi H, Doi Y, Liang J, Mizuno T, Suzumura A. Macrophage-induced neurotoxicity is mediated by glutamate and attenuated by glutaminase inhibitors and gap junction inhibitors. Life Sci. 2008. 82: 1111-6
280. Ye BS, Leung AOW, Wong MH. The association of environmental toxicants and autism spectrum disorders in children. Environ Pollut. 2017. 227: 234-42
281. Young AM, Campbell E, Lynch S, Suckling J, Powis SJ. Aberrant NF-kappaB expression in autism spectrum condition: A mechanism for neuroinflammation. Front Psychiatry. 2011. 2: 27-
282. Zaki MM, Abdel-Al H, Al-Sawi M. Assessment of plasma amino acid profile in autism using cation-exchange chromatography with postcolumn derivatization by ninhydrin. Turk J Med Sci. 2017. 47: 260-7
283. Zaky A, Mohammad B, Moftah M, Kandeel KM, Bassiouny AR. Apurinic/apyrimidinic endonuclease 1 is a key modulator of aluminum-induced neuroinflammation. BMC Neurosci. 2013. 14: 26-
284. Zhang Z, Bassam B, Thomas AG, Williams M, Liu J, Nance E. Maternal inflammation leads to impaired glutamate homeostasis and up-regulation of glutamate carboxypeptidase ii in activated microglia in the fetal/newborn rabbit brain. Neurobiol Dis. 2016. 94: 116-28
285. Zhang KL, Lou DD, Guan ZZ. Activation of the AGE/RAGE system in the brains of rats and in SH-SY5Y cells exposed to high level of fluoride might connect to oxidative stress. Neurotoxicol Teratol. 2015. 48: 49-55
286. Zhou Y, Danbolt NC. Glutamate as a neurotransmitter in the healthy brain. J Neural Transm. 2014. 121: 799-817
287. Zitka O, Skalickova S, Gumulec J, Masarik M, Adam V, Hubalek J. Redox status expressed as GSH: GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol Lett. 2012. 4: 1247-53

