

- Unit of Neurosurgery, S. Elia Hospital, Caltanissetta, Italy
- Department of Experimental Biomedicine and Clinical Neurosciences, School of Medicine, Neurosurgical Clinic, University of Palermo, Italy
- Unit of Pathology, S. Elia Hospital, Caltanissetta, Italy
Correspondence Address:
Rosario Maugeri
Unit of Neurosurgery, S. Elia Hospital, Caltanissetta, Italy
DOI:10.4103/sni.sni_434_17
Copyright: © 2018 Surgical Neurology International This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.How to cite this article: Luca Ruggeri, Nicola Alberio, Raffaele Alessandrello, Giovanni Cinquemani, Cesare Gambadoro, Rita Lipani, Rosario Maugeri, Francesco Nobile, Domenico Gerardo Iacopino, Giovanni Urrico, Roberto Battaglia. Rapid malignant progression of an intraparenchymal choroid plexus papillomas. 05-Jul-2018;9:131
How to cite this URL: Luca Ruggeri, Nicola Alberio, Raffaele Alessandrello, Giovanni Cinquemani, Cesare Gambadoro, Rita Lipani, Rosario Maugeri, Francesco Nobile, Domenico Gerardo Iacopino, Giovanni Urrico, Roberto Battaglia. Rapid malignant progression of an intraparenchymal choroid plexus papillomas. 05-Jul-2018;9:131. Available from: http://surgicalneurologyint.com/surgicalint-articles/rapid-malignant-progression-of-an-intraparenchymal-choroid-plexus-papillomas/
Date of Submission
09-Sep-2017
Date of Acceptance
29-May-2018
Date of Web Publication
05-Jul-2018
Abstract
Background:Choroid plexus tumors (CPTs) are rare neoplasms accounting for only 0.3–0.6% of all brain tumors in adults and 2–5% in children. The World Health Organization (WHO) classification describes three histological grades: grade I is choroid plexus papilloma (CPP), grade II is atypical papilloma, and grade III is the malignant form of carcinoma. In adults, CPTs rarely have a supratentorial localization.
Case Description:Here we report a very rare case of an intraparenchymal parietal CPP with a rapid histological transition from grade I to grade III WHO in a 67-year-old man, in
Conclusion:Because of the rarity of these oncotypes, descriptions of each new case are useful, mostly to consider this diagnostic entity in extraventricular brain tumors of adults, despite an unusual location.
Keywords: Choroid plexus atypical papilloma, choroid plexus carcinoma, choroid plexus papillomas, malignant progression, World Health Organization classification
BACKGROUND
Choroid plexus tumors (CPTs) are rare intracranial neoplasms that derive from the choroid plexus epithelium of ventricular system. CPTs account for only 0.3–0.6% of all brain tumors in adults and 2–5% in children.[
CPTs are of neuroectodermal origin, arising from epithelial cells of the choroid plexus and are found mostly in the lateral and fourth ventricles, and less commonly in the third ventricle.[
We report a very rare case of an intraparenchymal CPP with a documented rapid transition from benign to malignant histology.
CASE DESCRIPTION
A 67-year-old right-handed man presented to our department with a 4-month history of progressively worsening headaches and a 2-week history of slurred speech and lower limb weakness. On physical examination, the patient was alert and oriented but with a transcortical sensory aphasia associated with ideomotor apraxia and episodes of spatial disorientation. No other focal motor, sensory, or cerebellar abnormalities were detected, and the remainder of the physical examination was unremarkable. He was afebrile and had normal vital signs and normal findings of routine laboratory studies.
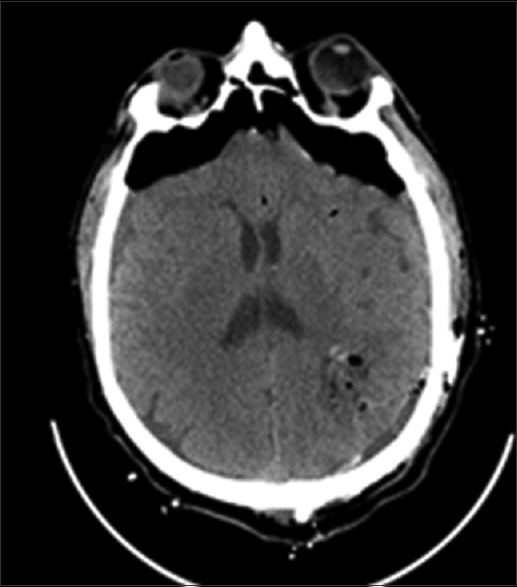
A computed tomography (CT) scan of the head showed a purely cystic mixed density parietal mass on left hemisphere. A contrast-enhanced magnetic resonance imaging (MRI) showed a minimally enhancing cyst wall, full of fluid, without a solid nodular component [
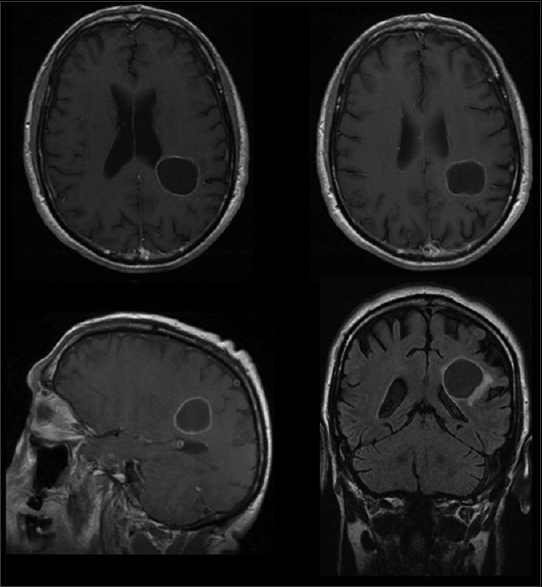
A left parietal craniotomy was performed. After the dura was opened, most of the cyst was removed and five soft-tissue samples specimens were sent for pathological analysis.
Feeding vessels were found deep near the area of the ventricular ependyma, but the ependyma of the ventricle was not breached during the operation. A partial excision of the cyst was performed, being the lesion in full eloquent area.
An autologous fibrin glue was used to enhance hemostasis and dural sealing.[
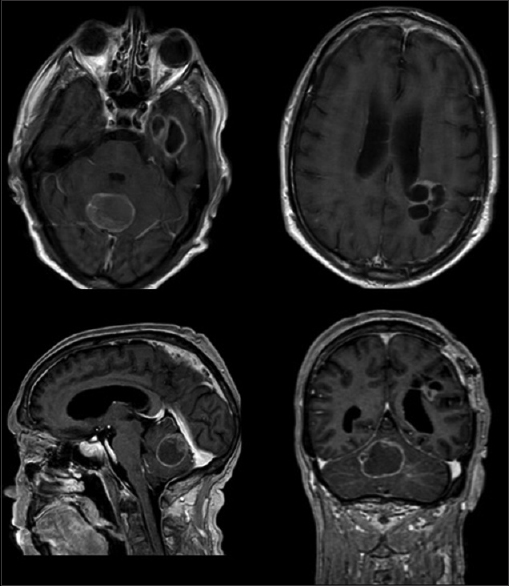
After 7 months the patient developed ataxia, dizziness, vomit, and worsening headache. Imaging showed significant regrowth of the known residual parietal tumor and a new cystic lesion in the cerebellar vermis and left fronto-basal supratentorial region [
Pathology
Gross examination of the fresh tumor specimen was performed on all available pathological samples (net weight of all the specimens: 5.5 g of size 4–12 mm). Histological diagnosis was assessed according to the WHO criteria for CPTs. CPP was composed of a delicate and vascularized connective tissue frond covered by a single papillary layer of uniform cuboidal to columnar epithelial cells with round or ovally, basally situated monomorphic nuclei. Mitotic activity was extremely low (<1 mitose per 10 hpfs). Brain invasion, increased cellular density, necrosis, and nuclear pleomorphism were absent. Immunohistochemically, CPP expressed cytokeratins. CK7/CK20 combination was CK7-positive and CK20-negative. Glial fibrillary acid protein (GFAP) was focally present. Benign CPP (WHO grade I°) was diagnosed.
The specimens from the second surgical procedure after 7 months were significant for multiple fragments of tan-pink, soft, and friable tissue collectively weighing 10.5 g and measuring 3 × 2, 5 × 2 cm. The tissue was fixed in 10% formalin and processed for paraffin embedding. The paraffin blocks were sectioned at 5-μm intervals and then stained with hematoxylin and eosin for microscopic examination. Histologically, malignant progression of CPP was reported. The tumor showed frank signs of malignancy. According to the WHO criteria, the surgical specimens showed frequent mitoses (>5 per 10 randomly selected hpfs), increased cellular density, nuclear pleomorphism, blurring of the papillary pattern with poorly structured sheets of tumor cells, and necrotic areas. The tumor strongly immunostained for cytokeratins and it was focally positive for GFAP. Immunohistochemical analysis of tissue proliferation was carried out using a monoclonal antibody against Ki67 (MIB-1, 1:100; DAKO) using the avidin–biotin method on an automated staining system. The differential diagnosis included papillary anaplastic ependymoma and metastatic carcinoma. Because the recurrence was characterized by frank signs of malignancy, according to the current WHO criteria, a diagnosis of CPC (WHO grade III) was made [
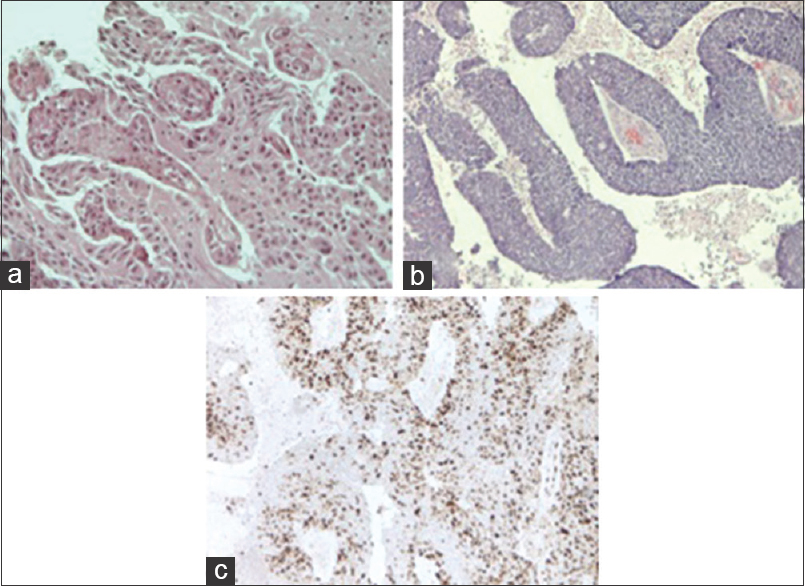
Figure 4
(a) Photomicrograph showing benign features of the CPP [hematoxylin and eosin (H and E) original magnification ×200] in the sample of the first surgical procedure. (b) Photomicrograph showing frank signs of malignancy of CPC (H and E original magnification ×100) in the sample of the second surgical procedure. (c) Demonstrates a representative area of the neoplasia with a high mitotic index (Ki-67 Ab, original magnification ×100)
DISCUSSION
Here, we report a very rare case of an intraparenchymal CPP with an early transition from grade I to grade III WHO in <7 months. Histological grading is recognized as an important prognostic factor in CPTs and also affects the decision toward adjuvant radiotherapy and chemotherapy.
In contrast to benign CPPs (WHO grade I), CPCs (WHO grade III) are characterized by frank signs of malignancy with frequent mitoses, nuclear pleomorphism, increased cellular density, blurring of the papillary growth pattern, and necrosis. The distinction between CPP versus CPC has not been always clear and some tumors showed only one or few histological features of malignancy as pleomorphism, increased mitotic activity, or invasion. Those tumors have been retrospectively termed atypical and classified in WHO grading as II.[
A study by Jeibmann et al. documented a series of 124 previously diagnosed CPPs, whose 15% were atypical. Author suggested that previous documented transition from benign to malignant histology might be overestimated, being extremely rare, instead of a more common evolution from grade II to the malignant form.[
In our case the first specimen revealed an extremely low mitotic activity (<1 mitose per 10 hpfs), absence of brain invasion, increased cellular density, necrosis, and nuclear pleomorphism. Immunohistochemically, there were no signs of atopy or malignancy so that a benign CPP (grade I WHO) was diagnosed.
After 7 months a new specimen in another surgical location revealed frank signs of malignancy. Frequent mitoses (>5 per 10 randomly selected hpfs), increased cellular density, nuclear pleomorphism, blurring of the papillary pattern with poorly structured sheets of tumor cells, and necrotic areas were documented. Immunohistochemical analysis revealed a high Ki67. So, a diagnosis of CPC (WHO grade III) was made.
To the best of our knowledge, this is the first reported case of a CPP arising in an extraventricular and extrachoroidal location which recurred, turning directly in malignant histology in few months. As known, in adults, CPT originates primarily in the fourth ventricle and only very rarely a localization dissociates to the ventricles or the choroid plexus.
Several theories have been proposed to explain the reasons why these lesions originated in such unusual locations. These lesions might arise from the embryonic residuals of ectopic secretory choroid plexus epithelium or, according another theory, ependymal tissue might become segregated during the development of the brain. Many ectopic CPTs are in proximity to the ventricular system, therefore these lesions possibly might arise within the nearby normal choroid plexus and be separated from it later as the tumor evolves. However, the exact etiopathogenetic mechanism remains unclear.[
In our case an attachment to the normal choroid plexus could not be identified at neuroimaging and the left lateral ventricle was effaced but not seemed involved with the tumor. Also after 6 months, following the rapid malignant evolution of the tumor, there was not an involvement of the ventricular area. Besides this, during the first surgical procedure, feeding vessels were found deep near the area of the ventricular ependyma, but the ependyma of the ventricle was not breached during the operation, looking as an intact and “healthy” layer.
According to our literature review only 9 cases of extraventricular supratentorial CPTs have been described in the literature: two pediatric cases with a CPT grade III, while the other reports have been described in adult patients (1 carcinoma, 2 atypical, 3 benign papillomas, and another case not specified).[
Malignant progression from papilloma to carcinoma is rare. Eight patients have been described in the literature to our knowledge. Jeibmann et al. reviewed these patients and stated that at least four are probably atypical papilloma recurring rather than a progression to carcinoma. Thus, the true incidence of malignant progression of CPP to CPC may be overestimated.[
CPPs are generally benign tumors, with reported 5-year overall survival (OS) rates of 90–100% and it is widely accepted that their complete excision is curative. Recurrence of CPP after gross total excision is reported, but uncommon. Jeibmann et al. found a recurrence rate of 6% for benign CPP and 29% for atypical CPP. Metastases of benign CPP are also rare and a review by Jinhu et al. identified only 14 well-documented patients.[
In our case, we performed a subtotal removal of the tumor for saving the near eloquent area. The situation is different for CPCs. CPCs carry a correspondingly worse prognosis; reported 5-year OS rates range from 26 to 41%.[
The present case emphasizes the importance of considering this diagnostic entity in an extraventricular brain tumor of adults despite an unusual location. We think that a closer neuroimaging follow-up might be advisable, although a diagnosis of grade I, being this hystotype seldom, also if very rarely, characterized by rapid malignant progression.
CONCLUSION
CPC is a rare diagnosis in adults, mostly if extraventricular. To the best of our knowledge, this is the first reported case of a CPP arising in an extraventricular and extrachroidal location which recurred, turning directly into malignant histology in few months. Nowadays, the natural history of this neoplasm is unknown. An improved understanding of the pathogenesis of these tumors will help clinicians in future diagnosis and treatment.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
We would like to thank Livia and Giuliano Maugeri for the help and the patience during the draft of the Manuscript.
References
1. Bahar M, Hashem H, Tekautz T, Worley S, Tang A, de Blank P. Choroid plexus tumors in adult and pediatric populations: The Cleveland Clinic and University Hospitals experience. J Neurooncol. 2017. 132: 427-32
2. Bettegowda C, Adogwa O, Mehta V, Chaichana KL, Weingart J, Carson BS. Treatment of choroid plexus tumors: A 20-year single institutional experience. J Neurosurg Pediatr. 2012. 10: 398-405
3. Cannon DM, Mohindra P, Gondi V, Kruser TJ, Kozak KR. Choroid plexus tumor epidemiology and outcomes: Implications for surgical and radiotherapeutic management. J Neurooncol. 2015. 121: 151-7
4. Carter AB, Price DL, Tucci KA, Lewis GK, Mewborne J, Singh HK. Choroid plexus carcinoma presenting as an intraparenchymal mass. J Neurosurg. 2001. 95: 1040-4
5. Dhillon RS, Wang YY, McKelvie PA, O’Brien B. Progression of choroid plexus papilloma. J Clin Neurosci. 2013. 20: 1775-8
6. Giugno A, Maugeri R, D’Arpa S, Visocchi M, Iacopino DG. Complex reconstructive surgery following removal of extra-intracranial meningiomas, including the use of autologous fibrin glue and a pedicled muscle flap. Interdisciplinary Neurosurgery: Advanced Techniques and Case Management. 2014. 1: 84-7
7. Jaiswal S, Vij M, Mehrotra A, Kumar B, Nair A, Jaiswal AK. Choroid plexus tumors: A clinico-pathological and neuro-radiological study of 23 cases. Asian J Neurosurg. 2013. 8: 29-35
8. Jeibmann A, Hasselblatt M, Gerss J, Wrede B, Egensperger R, Beschorner R. Prognostic implications of atypical histologic features in choroid plexus papilloma. J Neuropathol Exp Neurol. 2006. 65: 1069-73
9. Jeibmann A, Wrede B, Peters O, Wolff JE, Paulus W, Hasselblatt M. Malignant progression in choroid plexus papillomas. J Neurosurg. 2007. 107: 199-202
10. Jinhu Y, Jianping D, Jun M, Hui S, Yepeng F. Metastasis of a histologically benign choroid plexus papilloma: Case report and review of the literature. J Neurooncol. 2007. 83: 47-52
11. Lam S, Lin Y, Cherian J, Qadri U, Harris DA, Melkonian S. Choroid plexus tumors in children: A population-based study. Pediatr Neurosurg. 2013. 49: 331-8
12. Longatti P, Basaldella L, Orvieto E, Dei Tos A, Martinuzzi A. Aquaporin(s) expression in choroid plexus tumours. Pediatr Neurosurg. 2006. 42: 228-33
13. Lozier AP, Arbaje YM, Scheithauer BW. Supratentorial, extraventricular choroid plexus carcinoma in an adult: Case report. Neurosurgery. 2009. 65: E816-7
14. Maugeri R, Basile L, Giugno A, Graziano F, Iacopino DG. Impasse in the management of recurrent basal cell carcinoma of the skull with sagittal sinus erosion1. Interdisciplinary Neurosurgery: Advanced Techniques and Case Management. 2015. 2: 160-3
15. Maugeri R, Giammalva GR, Graziano F, Iacopino DG. May autologue fibrin glue alone enhance ossification?. An unexpected spinal fusion. World Neurosurg. 2016. 95: 611-2
16. Maugeri R, Schiera G, Di Liegro CM, Fricano A, Iacopino DG, Di Liegro I. Aquaporins and brain tumors. Int J Mol Sci. 2016. 17: E1029-
17. McCall T, Binning M, Blumenthal DT, Jensen RL. Variations of disseminated choroid plexus papilloma: 2 case reports and a review of the literature. Surg Neurol. 2006. 66: 62-7
18. Passariello A, Tufano M, Spennato P, Quaglietta L, Verrico A, Migliorati R. The role of chemotherapy and surgical removal in the treatment of choroid plexus carcinomas and atypical papillomas. Childs Nerv Syst. 2015. 31: 1079-88
19. Qi Q, Ni S, Zhou X, Huang B, Li X. Extraventricular intraparenchymal choroid plexus tumors in cerebral hemisphere: A series of 6 cases. World Neurosurg. 2015. 84: 1660-7
20. Stevens EA, Stanton CA, Nichols K, Ellis TL. Rare intraparenchymal choroid plexus carcinoma resembling atypical teratoid/rhabdoid tumor diagnosed by immunostaining for INI1 protein. J Neurosurg Pediatr. 2009. 4: 368-71
21. Turkoglu E, Kertmen H, Sanli AM, Onder E, Gunaydin A, Gurses L. Clinical outcome of adult choroid plexus tumors: Retrospective analysis of a single institute. Acta Neurochir (Wien). 2014. 156: 1461-8

