

- Department of Neurological Surgery, University of Texas Health Science Center, San Antonio, Texas, USA
- Department of Pathology, University of Texas Health Science Center, San Antonio, Texas, USA
- Department of Radiology, University of Texas Health Science Center, San Antonio, Texas, USA
Correspondence Address:
Derek C. Samples
Department of Neurological Surgery, University of Texas Health Science Center, San Antonio, Texas, USA
DOI:10.4103/2152-7806.195583
Copyright: © 2016 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Derek C. Samples, James Henry, Carlos Bazan, Izabela Tarasiewicz. A case of astroblastoma: Radiological and histopathological characteristics and a review of current treatment options. 12-Dec-2016;7:
How to cite this URL: Derek C. Samples, James Henry, Carlos Bazan, Izabela Tarasiewicz. A case of astroblastoma: Radiological and histopathological characteristics and a review of current treatment options. 12-Dec-2016;7:. Available from: http://surgicalneurologyint.com/surgicalint_articles/a-case-of-astroblastoma-radiological-and-histopathological-characteristics-and-a-review-of-current-treatment-options/
Date of Submission
10-Jun-2016
Date of Acceptance
17-Oct-2016
Date of Web Publication
12-Dec-2016
Abstract
Background:Astroblastoma is a rare neuroepithelial tumor that often originates in the cerebral hemisphere of children and young adults. Diagnosis of this obscure neoplasm can be difficult because these tumors are so infrequently encountered and share common radiological and neuropathological features of other glial neoplasms. As such, it should be included in the differential diagnosis of astrocytoma and ependymoma if the clinical and radiographic features suggest it. Standardized treatment of astroblastomas remains under dispute because of the lack of knowledge regarding the tumor and a paucity of studies in the literature.
Case Description:We present a case of a low-grade astroblastoma diagnosed in a 30-year-old female with seizures, headache, and vision changes. She underwent gross total resection and, without evidence of high-grade features, adjuvant therapy was not planned postoperatively. Post-operative surveillance suggested early recurrence, warranting referral to radiation therapy. Patient ended up expiring despite adjuvant therapy secondary to extensive recurrence and tumor metastasis.
Conclusions:Astroblastoma must be considered in the differential of supratentorial tumors in children and young adults. Treatment of such, as suggested by most recent literature, includes gross total resection and adjuvant radiotherapy for lesions exhibiting high-grade features.
Keywords: Adjuvant radiotherapy, astroblastoma, brain edema, case report, cerebrum
INTRODUCTION
First described by Bailey and Cushing in 1926, astroblastomas are rare glial tumors, accounting for approximately 0.4–2.8% of primary brain tumors.[
CASE REPORT
A 30-year-old woman presented with a seizure, right scalp paresthesias, a right-sided headache, blurred vision, and nausea. Her neurological examination was unremarkable, however, formal ophthalmological testing demonstrated left homonymous quadrantanopsia. She underwent a non-contrast computed tomography (CT), with results indicating an intracranial neoplasm. Subsequently, magnetic resonance imaging (MRI) of the brain with intravenous (IV) gadolinium was obtained, which demonstrated a 6.1 cm heterogeneously enhancing mass in the posteromedial aspect of the right temporoparietal lobes protruding into the right atrium. It displayed mixed T1 and T2 signal intensity with regions of restricted diffusion. There was mild peritumoral T2 hyperintensity, signifying peritumoral edema. A large area of gradient susceptibility along the posterior aspect of the lesion represented calcifications [
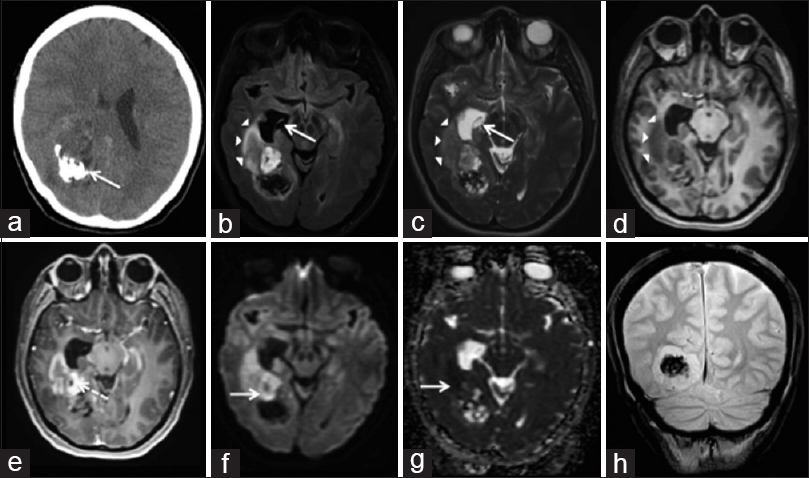
Figure 1
Astroblastoma – Axial NECT (a) shows a heterogenous mass in the right temporal lobe with calcification (arrow). Axial FLAIR (b), T2W (c), and T1W pre-contrast (d) images show T1 & T2 prolongation in the right temporal lobe (arrowheads) and right temporal horn. There is dilation of the right temporal horn (arrow). The T1W post-contrast image (e) shows lesion enhancement and enhancement of cysts (dotted arrow). There is diffusion restriction (arrow) on DWI (f) and ADC (g). The coronal gradient T2* image (h) shows a region of susceptibility artifact compatible with calcification.
The patient subsequently underwent a right craniotomy for biopsy and partial resection, with initial pathology described as a low-grade astrocytic tumor. She continued to be symptomatic with headaches and underwent complete resection 2 months later; the macrocalcification was left behind due to tethering of normal brain tissue. Postoperatively, she had persistent resection cavity fluid collections, as well as a trapped right temporal horn. She subsequently underwent cystoperitoneal shunt placement. Postoperatively, she required one proximal revision of her shunt secondary to malfunction, but otherwise remained clinically stable with resolution of her headaches and no additional neurological deficits. Three months after the resection, imaging follow-up demonstrated interval enhancement of the surgical cavity, suggestive of local recurrence. Histologically, the tumor demonstrated epithelioid cells with short cytoplasmic processes arranged in perivascular pseudorosettes. Minimal mitotic activity was observed with PHH3, and no high-grade features were identified. The neoplasm was positive for glial fibrillary acidic protein (GFAP), synaptophysin, and Olig2; IDH1 was negative, similar to the staining profiles reported in recent literature [
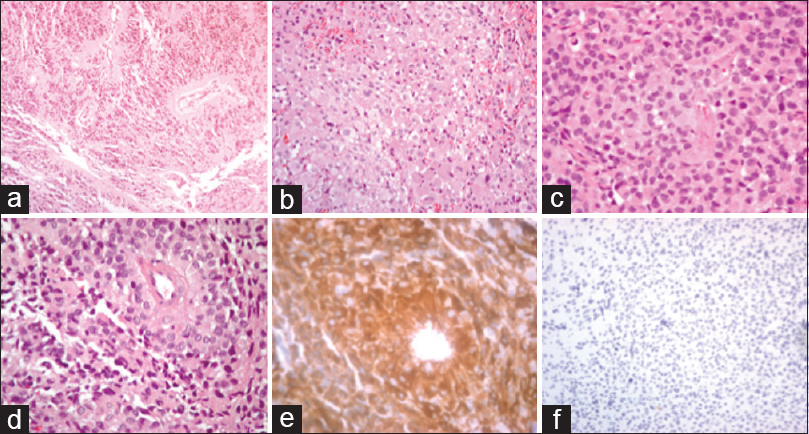
Figure 2
Astroblastoma Immunohistochemistry – ×100, ×200, and ×400 hematoxylin and eosin (H&E) staining depicting epithelioid cells of the astroblastoma (a-c). ×400 H&E staining demonstrating astroblastoma cells with short cytoplasmic processes arranged in perivascular pseudorosettes (d). ×400 GFAP+ staining (e). ×200 PHH3 positive staining at (f)
DISCUSSION
In the most recent updated WHO classification of CNS tumors, astroblastomas are classified in the category of “other neuroepithelial tumors,” formerly designated as neuroepithelial tumors of uncertain origin.[
Demographics can be helpful in differentiating astroblastoma from other tumors. A bimodal distribution is commonly observed, with the majority of reported cases occurring in patients between 10 to 30 years of age.[
Imaging findings can offer additional clues to support the diagnosis and prognosis. Astroblastomas often demonstrate T1 and T2-prolongation relative to white matter, with well-demarcated boundaries and heterogeneous contrast enhancement.[
As a consequence of their extreme rarity, comprising a few hundred reported cases, astroblastomas present a challenge in terms of diagnosis and selection of appropriate treatment.[
Certain indices suggestive of high grade/malignant lesions include the extent of peritumoral T2 signal on MRI, cytological atypia, high Ki-67 labeling index, tumor necrosis, increased cellularity, and vascular proliferation.[
CONCLUSIONS
When encountering a well-demarcated supratentorial mass with heterogeneous enhancement and macrocalcifications in a child or young adult, astroblastoma should be considered in the differential diagnosis. Based on the micro/macroscopic features, clinicians should address this tumor more along the lines of ependymoma with respect to treatment, with gross total resection being the primary goal. In the same manner, despite the lack of standardized treatment protocols, the literature indicates that gross total resection provides the best outcome for low grade lesions, which can be followed closely with imaging, while adjuvant radiotherapy should be offered for high risk patients presenting with significant peritumoral preoperative edema on T2 sequences and high grade histopathological features. However, close follow-up is recommended because tumors with low-grade features have been shown to recur and warrant additional treatment.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Ahmed K, Allen P, Mahajan A, Brown P, Ghia A. Astroblastomas: A Surveillance, Epidemiology, and End Results (SEER)-Based Patterns of Care Analysis. World Neurosurg. 2014. 82: e291-7
2. Agarwal V, Mally R, Palande DA, Velho V. Cerebral astroblastoma: A case report and review of literature. Asian J Neurosurg. 2012. 7: 98-100
3. Bell JW, Osborn AG, Salzman KL, Blaser SI, Jones BV, Chin SS. Neuroradiologic characteristics of astroblastoma. Neuroradiology. 2007. 49: 203-9
4. Binesh F, Akhavan A, Navabii H, Mehrabaniyan M. Anaplastic astroblastoma: A rare glial tumour. BMJ Case Rep 2011. 2011. p.
5. Bonnin J, Rubinstein L. Astroblastomas: A pathological study of 23 tumors, with postoperative follow-up in 13 patients. Neurosurgery. 1989. 25: 6-13
6. Brat DJ, Hirose Y, Cohen KJ, Feuerstein BG, Burger PC. Astroblastoma: Clinicopathologic features and chromosomal abnormalities defined by comparative genomic hybridization. Brain Pathol Zurich Switz. 2000. 10: 342-52
7. Denaro L, Gardiman M, Calderone M, Rossetto M, Ciccarino P, Giangaspero F. Intraventricular astroblastoma. Case report. J Neurosurg Pediatr. 2008. 1: 152-5
8. Fu YJ, Taniguchi Y, Takeuchi S, Shiga A, Okamoto K, Hirato J. Cerebral astroblastoma in an adult: An immunohistochemical, ultrastructural and genetic study. Neuropathology. 2013. 33: 312-9
9. Fuller GN, Scheithauer BW. The 2007 Revised World Health Organization (WHO) Classification of Tumours of the Central Nervous System: Newly codified entities. Brain Pathol. 2007. 17: 304-7
10. Ganapathy S, Kleiner LI, Mirkin DL, Broxson E. Unusual manifestations of astroblastoma: A radiologic-pathologic analysis. Pediatr Radiol. 2009. 39: 168-71
11. Janz C, Buhl R. Astroblastoma: Report of two cases with unexpected clinical behavior and review of the literature. Clin Neurol Neurosurg. 2014. 125: 114-24
12. Lau P, Thomas T, Lui P, Khin A. Low grade astroblastoma with rapid recurrence: A case report. Pathology. 2006. 38: 78-80
13. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007. 114: 97-109
14. Mangano FT, Bradford AC, Mittler MA, Valderrama E, Schneider SJ. Astroblastoma. Case report, review of the literature, and analysis of treatment strategies. J Neurosurg Sci. 2007. 51: 21-7
15. Narayan S, Kapoor A, Singhal MK, Jakhar SL, Bagri PK, Rajput PS. Astroblastoma of cerebrum: A rare case report and review of literature. J Can Res Ther. 2015. 11: 667-
16. Notarianni C, Akin M, Fowler M, Nanda A. Brainstem astroblastoma: A case report and review of the literature. Surg Neurol. 2008. 69: 201-5
17. Port JD, Brat DJ, Burger PC, Pomper MG. Astroblastoma: Radiologic-pathologic correlation and distinction from ependymoma. AJNR Am J Neuroradiol. 2002. 23: 243-7
18. Salvati M, D’Elia A, Brogna C, Frati A, Antonelli M, Giangaspero F. Cerebral astroblastoma: Analysis of six cases and critical review of treatment options. J Neurooncol. 2009. 93: 369-78
19. Shen F, Chen L, Yao Y, Zhou L. Astroblastoma: Rare incidence and challenges in the pattern of care. World Neurosurg. 2014. 82: e125-7
20. Sughrue M, Choi J, Rutkowski M, Aranda D, Kane A, Barani I. Clinical features and post-surgical outcome of patients with astroblastoma. J Clin Neurosci. 2011. 18: 750-4
21. Yao K, Bin W, Xi B, Xi M, Duan Z, Wang J, Qi X. Distant dissemination of mixed low-grade astroblastoma-arteriovenous malformation after initial operation: A case report. Int J Clin Exp Pathol. 2015. 8: 7450-6

