

- Department of Neurosurgery, Hospital General de Culiacán, Salvador Zubirán, México
- Department of Anatomical Pathology, Hospital General de Culiacán, Salvador Zubirán, México
- Department of Anatomical Pathologyl, Hospital Angeles de Culiacán Sinaloa, Salvador Zubirán, México
- Department of Anatomical Pathology, Instituto Nacional de Ciencias Medicas y Nutrición, Salvador Zubirán, México
Correspondence Address:
Jesús Rocha-Maguey
Department of Anatomical Pathology, Instituto Nacional de Ciencias Medicas y Nutrición, Salvador Zubirán, México
DOI:10.4103/2152-7806.175070
Copyright: © 2016 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Jesús Rocha-Maguey, José-Angel Felix-Torrontegui, Myriam Cabrera-López, Macrina Gutiérrez-Castro, de Oca DM. A new case of cervical intramedullary sinus histiocytosis causing paraplegia and review of the literature. Surg Neurol Int 28-Jan-2016;7:9
How to cite this URL: Jesús Rocha-Maguey, José-Angel Felix-Torrontegui, Myriam Cabrera-López, Macrina Gutiérrez-Castro, de Oca DM. A new case of cervical intramedullary sinus histiocytosis causing paraplegia and review of the literature. Surg Neurol Int 28-Jan-2016;7:9. Available from: http://surgicalneurologyint.com/surgicalint_articles/a-new-case-of-cervical-intramedullary-sinus-histiocytosis-causing-paraplegia-and-review-of-the-literature/
Abstract
Background:Rosai–Dorfman disease (RDD) is an uncommon, benign histiocytic proliferative disorder of unknown origin. It predominantly affects the lymph nodes, but can also be found extranodal in different organs. Nervous system involvement is rare, and the most cases are intracranial. Surgical treatment is indicated when the central nervous system (CNS) in compromised.
Case Description:We herein describe the management of a 27-year-old woman who presented progressive spinal cord symptoms, secondary to an isolated intramedullary lesion, which had a histological confirmation of RDD. To our knowledge, this is the 6th case reported in English written manuscripts. We review these cases and analyze some of the literature concerning the disease.
Conclusions:RDD shows some variability in the involvement of the entire neuraxis, and because its ability to mimic meningeal and primary brain tumors, it is essential to be aware of this entity and consider RDD in the differential diagnosis of various lesions of the CNS. The conclusive diagnosis must be obtained by histological methods, so surgical approaches have to be discussed. Although it is not considered as a malignancy, options for postoperative medical treatment are variable and include radiation, chemotherapy or maybe monoclonal antibodies for refractory or recurrent cases.
Keywords: Cervical, Rosai–Dorfman disease, sinus histiocytosis, spinal cord
INTRODUCTION
Sinus histiocytosis with massive lymphadenopathy (SHML), initially described in 1969 by Rosai and Dorfman,[
CASE PRESENTATION
This 27-year-old woman was in good health until 2 months before the discovery of her disease. On May 2014, she felt down spontaneously while walking. After this incident, she noticed certain difficulties in controlling her legs. Two days later, a progressive strength loss was developed on both legs, leaving her in a wheelchair 1 week later. Sphincters were intact at this time.
She consulted a general physician, who prescribed corticoid medication, obtaining some relief of her symptoms but never a complete recovery. After 6 weeks, she developed a complete and symmetrical spinal cord syndrome, referring an important weakness and numbness of the fingers on both hands and extending to the rest of her body. At this point, she developed urinary incontinence and severe constipation.
During the general examination, the patient was alert and cooperative. No fever present and we did not discover any important information on the pulmonary nor cardiovascular condition, no lymph nodes were palpated, the skin showed normal characteristics. The neurological exam demonstrated complete motor loss of both legs, with a partial sensory level of hypesthesia and hypalgesia at C6–C7. Reflexes were all hyperactive, with a positive bilateral Hoffman reflex and bilateral extensor plantar response. Laboratory data were entirely normal. A moderate increase on the erythrocyte sedimentation rate of 25 mm/h (normal: 15 mm/h) was detected. The cerebrospinal fluid analysis was within limits, with negative cultures. Magnetic resonance images (MRIs) of the cervical spine [
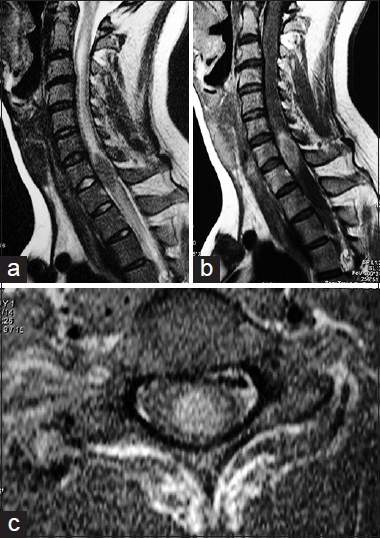
Figure 1
Magnetic resonance images of the cervical spine demonstrating a well-defined oval intramedullary lesion expanding the spinal cord at C7–Th1. (a) Sagittal T2-weighted-images demonstrating a well-defined hypointense mass causing an important edema above and under it. (b) Homogenous gadolinium enhancement is evident. (c) A predominant anterior location within the spinal cord is observed on axial images after the application of gadolinium
The patient was taken into surgery, where we performed a C6–Th1 laminoplasty. The lesion was identified using transoperative ultrasonogram. After opening the dura, we observed a severely expanded spinal cord, but with a normal superficial appearance. A midline myelotomy disclosed a well-defined brownish soft mass that was progressively dissected from the spinal cord parenchyma and achieving an in bloc resection with the aid of bipolar forceps and microscissors.
The patient had an uncomplicated postoperative evolution. The histology of the lesion [
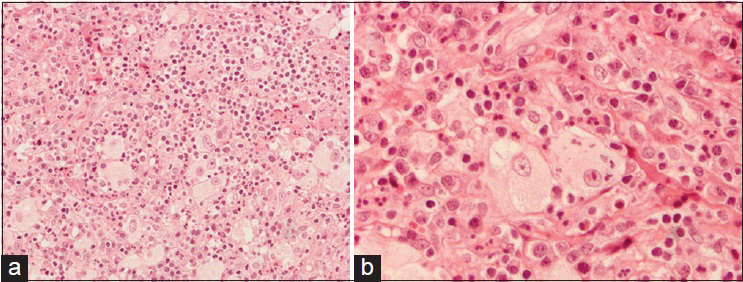
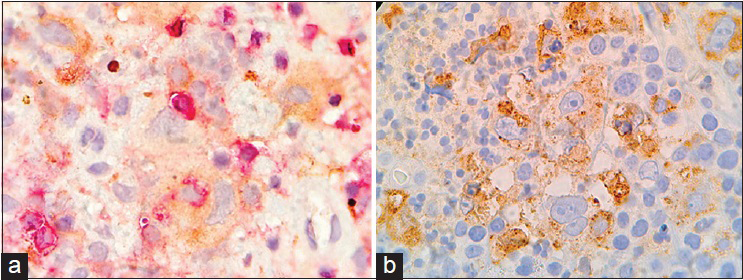
DISCUSSION
The non-Langerhans cells histiocytosis are thought to arise from either a dendritic cell or a macrophage cell line, and can be clinically divided into three major groups – those that primarily affect the skin, such as the juvenile xantogranuloma (JXG) family, and reticulohistiocytoma; those that affect the skin, but have a major systemic component such as xanthoma disseminatum and multicentric reticulohistiocytosis; and those that predominantly involve systemic sites, although skin may also be affected, such as systemic JXG and sinus histiocytosis with massive lymphadenopathy (SHML) or Rosai–Dorfman disease (RDD).[
The first report of SHML was described in 1965, by Destombes, as a lipid storage disorder developing after inflammation.[
RDD of the nervous system has also been reported in <5% of all cases and most of them in the brain.[
Rosai and Dorfman[
The current hypothesis is that RDD is a subtle, yet undefined immunological defect, which promotes monocyte recruitment from the circulation into the nodal or extranodal sites, followed by transformation into the immunophenotypically distinct RDD histiocytes, demonstrating emperipolesis with functional uniqueness in terms of the cytokine expression profile.[
However, evidence of RDD exclusive to the spine is increasing in the literature, with symptoms typical to the affected level, including weakness and sensory changes; most of them have been reported with an epidural or subdural location.
Over 90% of the CNS Rosai–Dorfman involve the leptomeninges and are observed on neuroimaging as a dural-based, contrast-enhancing lesion, and mimicking a meningioma.[
We elected to perform a three-level laminoplasty because no spinal instability was identified; the excision of the lesion was controlled under the sonographic image, assuming that the lesion was a primary ependymal tumor. Contrary to other authors, we detected a very well-defined plane of cleavage between the lesion and the spinal cord, making a complete excision very suitable, although the consistency of the lesion was very similar to that of a neurinoma. Besides of the preoperative clinical manifestations, there were non-neurologic complications and a progressive improvement was evident. Whatever the approach, surgical removal has been considered to be as the primary mode of treatment of CNS RDD in most of the reported cases, where cerebral or spinal decompression is the gold standard. Irradiation and/or steroid treatment have been tried in several cases with variable results. In one instance of RDD with spinal involvement, dramatic remission between prednisolone and vinblastine, without surgery, was mentioned.[
Since results with chemotherapy for RDD have not been encouraging, the use of chemotherapy is restricted to patients with the life-threatening disease or multiple relapses. Imatinib, a platelet-derived growth factor receptor B-inhibitor, demonstrated acceptable results in a recent case with RDD.[
CONCLUSIONS
The CNS involvement in RDD in various presentations is considered to be rare though there is an increase in the number of cases that have been documented of CNS-RDD. However, because of the variability of the involvement of the entire neuraxis, and its ability to mimic meningeal and primary brain tumors, it is essential to be aware of this entity and consider RDD in the differential diagnosis of various lesions of the CNS. The conclusive diagnosis must be obtained by histological methods. Since RDD was first described over 60 years ago, the mechanism behind this disease has not been discovered. Since it cannot be associated with a direct monoclonal cell population abnormality, RDD should not be considered as a malignancy, and it is also uncertain that an infectious or immunologic process may be responsible. Isolated molecular studies have suggested that RDD can be a polyclonal disorder. Intraxial presentation of RDD should be considered when a well-defined nodule surrounded by vasogenic edema is identified via radiologic examination. And finally, laminoplasty is a very well-developed procedure all around the world that we recommend for similar cases, because spinal stability is preserved, and with the aid of complementary ultrasonic aspiration, better chances to preserve neurological function will exist. The outcome is generally good, and the disease is usually self-limited; however, approximately 5–11% of patients die from this disease. Patients with combined immunologic abnormalities have a less favorable outcome and a higher fatality rate.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Abou-Zeid AH, Herwadkar A, du Plessis D, Gnanalingham KK. Isolated extradural Rosai-Dorfman disease of the thoracic spine: A rare cause of spinal cord compression: Case report. Neurosurgery. 2010. 67: E514-E515
2. Andriko JA, Morrison A, Colegial CH, Davis BJ, Jones RV. Rosai-Dorfman disease isolated to the central nervous system: A report of 11 cases. Mod Pathol. 2001. 14: 172-8
3. Becroft DM, Dix MR, Gillman JC, MacGregor BJ, Shaw RL. Benign sinus histiocytosis with massive lymphadenopathy: Transient immunological defects in a child with mediastinal involvement. J Clin Pathol. 1973. 26: 463-9
4. Cooper SL, Jenrette JM. Rosai-Dorfman disease: Management of CNS and systemic involvement. Clin Adv Hematol Oncol. 2012. 10: 199-202
5. Destombes P. Adenitis with lipid excess, in children or young adults, seen in the Antilles and in Mali. (4 cases). Bull Soc Pathol Exot Filiales. 1965. 58: 1169-75
6. El Molla M, Mahasneh T, Holmes SE, Al-Khawaja D. Rare presentation of Rosai-Dorfman disease mimicking a cervical intramedullary spinal cord tumor. World Neurosurg. 2014. 81: 442e7-442e9
7. Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): Review of the entity. Semin Diagn Pathol. 1990. 7: 19-73
8. Haas RJ, Helmig MS, Prechtel K. Sinus histiocytosis with massive lymphadenopathy and paraparesis: Remission with chemotherapy. A case report. Cancer. 1978. 42: 77-80
9. Hargett C, Bassett T. Atypical presentation of sinus histiocytosis with massive lymphadenopathy as an epidural spinal cord tumor: A case presentation and literature review. J Spinal Disord Tech. 2005. 18: 193-6
10. Huang YC, Tan HY, Jung SM, Chuang WY, Chuang CC, Hsu PW. Spinal epidural Rosai-Dorfman disease preceding by relapsing uveitis: A case report with literature review. Spinal Cord. 2007. 45: 641-4
11. Jones MP, Rueda-Pedraza ME. Extranodal sinus histiocytosis with massive lymphadenopathy presenting as an intramedullary spinal cord tumor: A case report. Am J Hematol. 1997. 54: 253-7
12. Konca C, Ozkurt ZN, Deger M, Aki Z, Yagci M. Extranodal multifocal Rosai-Dorfman disease: Response to 2-chlorodeoxyadenosine treatment. Int J Hematol. 2009. 89: 58-62
13. Le Guenno G, Galicier L, Uro-Coste E, Petitcolin V, Rieu V, Ruivard M. Successful treatment with azathioprine of relapsing Rosai-Dorfman disease of the central nervous system. J Neurosurg. 2012. 117: 486-9
14. Ma J, Xiao J, Wang L. Extranodal Rosai-Dorfman disease with multilevel lumbar spinal lesions. J Neurosurg Spine. 2008. 9: 55-7
15. Mehraein Y, Wagner M, Remberger K, Füzesi L, Middel P, Kaptur S. Parvovirus B19 detected in Rosai-Dorfman disease in nodal and extranodal manifestations. J Clin Pathol. 2006. 59: 1320-6
16. Middel P, Hemmerlein B, Fayyazi A, Kaboth U, Radzun HJ. Sinus histiocytosis with massive lymphadenopathy: Evidence for its relationship to macrophages and for a cytokine-related disorder. Histopathology. 1999. 35: 525-33
17. Molina-Carrión LE, Mendoza-Álvarez SA, Vera-Lastra OL, Caldera-Duarte A, Lara-Torres H, Hernández-González C. Rosai-Dorfman disease with spinal and cranial tumors. A clinical case reported. Rev Med Inst Mex Seguro Soc. 2014. 52: 218-23
18. Osenbach RK. Isolated extranodal sinus histiocytosis presenting as an intramedullary spinal cord tumor with paraplegia. Case report. J Neurosurg. 1996. 85: 692-6
19. Paulli M, Bergamaschi G, Tonon L, Viglio A, Rosso R, Facchetti F. Evidence for a polyclonal nature of the cell infiltrate in sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Br J Haematol. 1995. 91: 415-8
20. Petschner F, Walker UA, Schmitt-Gräff A, Uhl M, Peter HH. “Catastrophic systemic lupus erythematosus” with Rosai-Dorfman sinus histiocytosis. Successful treatment with anti-CD20/rutuximab. Dtsch Med Wochenschr. 2001. 126: 998-1001
21. Raslan O, Ketonen LM, Fuller GN, Schellingerhout D. Intracranial Rosai-Dorfman disease with relapsing spinal lesions. J Clin Oncol. 2008. 26: 3087-9
22. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol. 1969. 87: 63-70
23. Sandoval-Sus JD, Sandoval-Leon AC, Chapman JR, Velazquez-Vega J, Borja MJ, Rosenberg S. Rosai-Dorfman disease of the central nervous system: Report of 6 cases and review of the literature. Medicine (Baltimore). 2014. 93: 165-75
24. Shuangshoti SH, Navalitloha Y, Sukpanichnant S, Unhasuta CH, Shuangshoti S. Central nervous system involvement in Rosai-Dorfman disease: Report of a case with a review of the literature. Neuropathology. 1999. 19: 341-6
25. Triana-Pérez AB, Sánchez-Medina Y, Pérez-Del Rosario PA, Millán-Corada AM, Gómez-Perals LF, Domínguez-Báez JJ. Isolated intracranial Rosai-Dorfman disease: A case report and literature review. Neurocirugia (Astur). 2011. 22: 255-60
26. Utikal J, Ugurel S, Kurzen H, Erben P, Reiter A, Hochhaus A. Imatinib as a treatment option for systemic non-Langerhans cell histiocytoses. Arch Dermatol. 2007. 143: 736-40
27. Vaiselbuh SR, Bryceson YT, Allen CE, Whitlock JA, Abla O. Updates on histiocytic disorders. Pediatr Blood Cancer. 2014. 61: 1329-35
28. Warrier R, Chauhan A, Jewan Y, Bansal S, Craver R. Rosai-Dorfman disease with central nervous system involvement. Clin Adv Hematol Oncol. 2012. 10: 196-8
29. Yao K, Li TF, Zhu MW, Duan ZJ, Hu ZL, Bian Y. An intramedullary cervical cord lesion in a 12-year-old girl. Neuropathology. 2013. 33: 582-5

