

- Department of Neurosurgery, Southern District Health Board, Dunedin, Otago, New Zealand.
DOI:10.25259/SNI_225_2021
Copyright: © 2021 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Stephen Yu, Ramakrishna Bethanabatla, Ahmed Taha. A case report of lymphocytic hypophysitis. 07-Jun-2021;12:263
How to cite this URL: Stephen Yu, Ramakrishna Bethanabatla, Ahmed Taha. A case report of lymphocytic hypophysitis. 07-Jun-2021;12:263. Available from: https://surgicalneurologyint.com/?post_type=surgicalint_articles&p=10864
Date of Submission
06-Apr-2021
Date of Acceptance
20-May-2021
Date of Web Publication
07-Jun-2021
Abstract
Background: Lymphocytic hypophysitis (LH) is a rare condition that mostly affects women of the reproductive age. Because it is infrequently encountered, it is not often considered as a differential diagnosis of sellar masses. The diagnosis is made clinically with the aid of magnetic resonance imaging (MRI) and should be considered if the patient has endocrine derangements in addition to a sellar mass.
Case Description: A 37-year-old female presents with a complaint of headaches and CT imaging showed a sellar mass. She was also being investigated simultaneously by the endocrine department and was diagnosed with panhypopituitarism. She proceeded to surgery for a presumed pituitary adenoma but histopathology returned as LH.
Conclusion: It is important to have a wide differential diagnosis when managing pituitary masses. Clinical correlation with atypical MRI findings is useful to determine the diagnosis of LH.
Keywords: Hypophysitis, Neuroendocrine, Neuroradiology
INTRODUCTION
Lymphocytic hypophysitis (LH) was first reported in 1962 and is the most common variant of autoimmune hypophysitis.[
Autoimmune hypophysitis is often classified according to histopathology with the other histological subtypes being granulomatous, xanthomatous, and plasmacytic.[
The estimated annual incidence of hypophysitis is 1 in 7–9 million and there have been over 390 cases reported of LH.[
Given the rarity of the condition, we would like to present what may be the first case of LH in New Zealand.
CASE REPORT
A 37-year-old female was referred to the neurology service with a complaint of headache for the past 4–5 months. The headaches were severe and distributed over the entirety of her head and associated with fatigue and weight gain. The patient’s only medical history was primary amenorrhea secondary to an imperforate hymen.
She was trialed on migraine medications and a CT head was done which showed a pituitary mass. A subsequent magnetic resonance imaging (MRI) scan described the pituitary gland as enlarged with diffuse enhancement consistent with a macroadenoma [
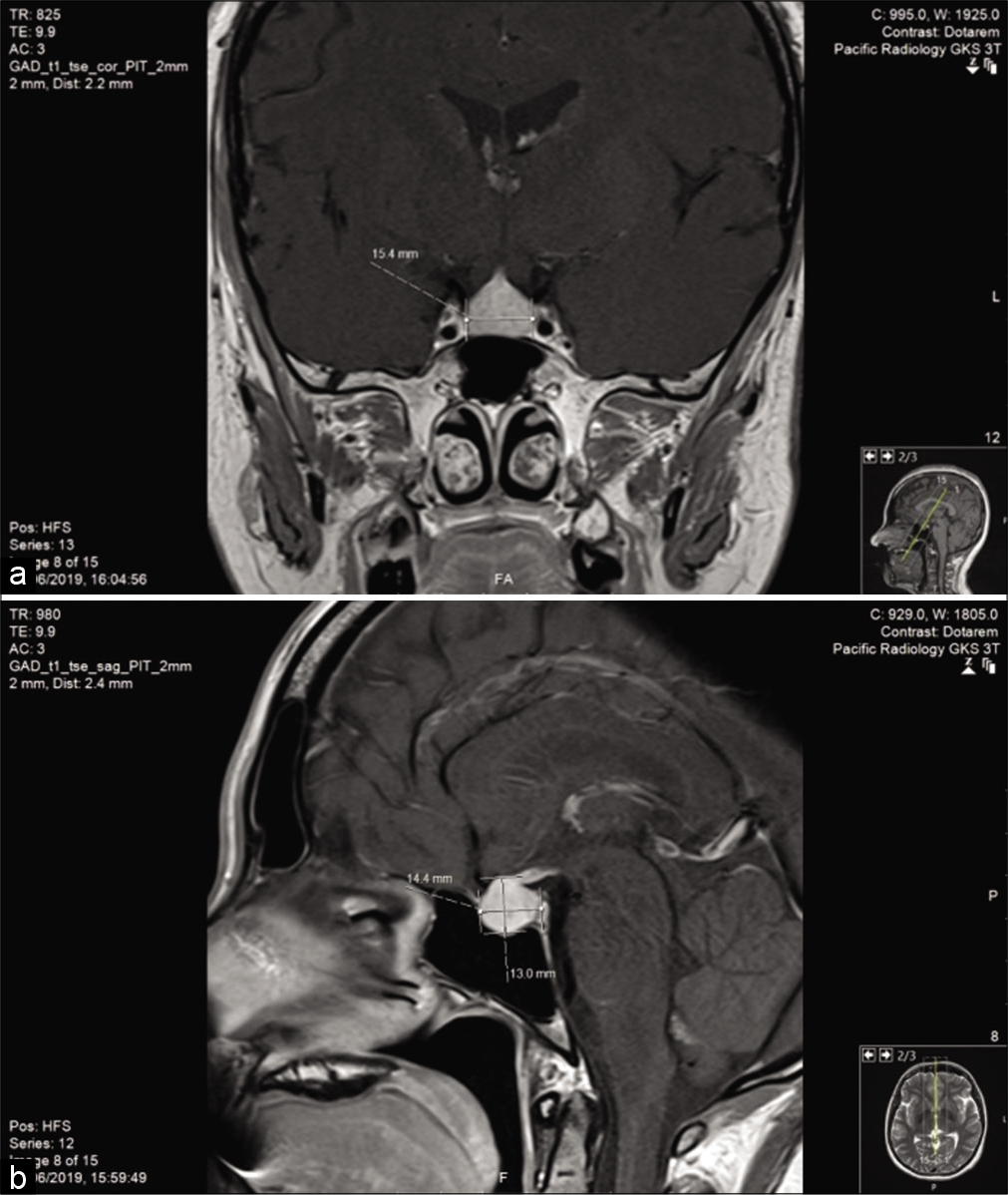
Simultaneously, the patient was being evaluated by endocrinology for deranged thyroid function tests. Further testing revealed a low cortisol level, LH, FSH, oestradiol, free T4, and TSH. She was diagnosed with hypopituitarism and treated with hydrocortisone and thyroxine.
The patient was referred to neurosurgery for resection of the pituitary lesion. This was done with a transsphenoidal approach and the surgery was uncomplicated. The histology returned as LH. The postoperative course was complicated by the development of diabetes insipidus, CSF leak requiring a return to theater and a readmission for meningitis. The patient is now recovering well on subsequent follow-up appointments.
DISCUSSION
LH is rare autoimmune disease and is challenging to diagnose. It predominantly affects women of the reproductive age and the most common symptom is persistent headache and endocrine deficiencies.[
Like other autoimmune diseases, the pathogenesis is due to the formation of autoantigens.[
MRI is the imaging modality of choice when investigating pituitary pathology, however, it is difficult to differentiate between LH and pituitary adenomas.[

Table 1:
Radiologic scoring system from Gutenberg et al.[
A limitation of Gutenberg et al.’s scoring system is that it has not been tested against other types of sellar masses as the study exclusively compared autoimmune hypophysitis to adenomas.[
In regard to treatment, it is important to note that no prospective controlled studies exist that have examined the treatment of hypophysitis.[
CONCLUSION
Our case highlights the difficulty of diagnosing and treating the rare condition of LH. It is important to have a high index of suspicion when presented with patients with intractable headaches, panhypopituitarism, and a sellar mass. Distinguishing LH from a pituitary adenoma is important due to the increasing evidence which suggests that medical management is the preferred first-line option for LH.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Caturegli P, Newschaffer C, Olivi A, Pomper MG, Burger PC, Rose NR. Autoimmune hypophysitis. Endocr Rev. 2005. 26: 599-614
2. Faje A. Hypophysitis: Evaluation and management. Clin Diabetes Endocrinol. 2016. 2: 15
3. Goudie RB, Pinkerton PH. Anterior hypophysitis and Hashimoto’s disease in a young woman. J Pathol Bacteriol. 1962. 83: 584-5
4. Gutenberg A, Larsen J, Lupi I, Rohde V, Caturegli P. A radiologic score to distinguish autoimmune hypophysitis from nonsecreting pituitary adenoma preoperatively. AJNR Am J Neuroradiol. 2009. 30: 1766-72
5. Honegger J, Buchfelder M, Schlaffer S, Droste M, Werner S, Strasburger C. Treatment of primary hypophysitis in Germany. J Clin Endocrinol Metab. 2015. 100: 3460-9
6. Iuliano SL, Laws ER. The diagnosis and management of lymphocytic hypophysitis. Expert Rev Endocrinol Metab. 2011. 6: 777-83
7. Kyriacou A, Gnanalingham K, Kearney T. Lymphocytic hypophysitis: Modern day management with limited role for surgery. Pituitary. 2007. 20: 241-50
8. Landek-Salgado MA, Gutenberg A, Lupi I, Kimura H, Mariotti S, Rose NR. Pregnancy, postpartum autoimmune thyroiditis, and autoimmune hypophysitis: Intimate relationships. Autoimmun Rev. 2010. 9: 153-7
9. Moskowitz SI, Hamrahian A, Prayson RA, Pineyro M, Lorenz RR, Weil RJ. Concurrent lymphocytic hypophysitis and pituitary adenoma. Case report and review of the literature. J Neurosurg. 2006. 105: 309-14

