

- Department of Medical, Surgical Sciences and Advanced Technologies “G.F. Ingrassia”, Anatomic Pathology, University of Catania,
- Department of Medical, Surgical Sciences and Advanced Technologies “G.F. Ingrassia”, Neurological Surgery, Policlinico “Rodolico-San Marco” University Hospital, University of Catania, 95123 Catania,
- Neuropathology Unit, Catholic University, Fondazione Policlinico Universitario “A. Gemelli” IRCSS, Rome, Italy.
Correspondence Address:
Giuseppe Broggi, Department “G.F. Ingrassia”, University of Catania, Catania, Italy.
DOI:10.25259/SNI_500_2021
Copyright: © 2021 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Giuseppe Broggi1, Francesco Certo2, Roberto Altieri2, Rosario Caltabiano1, Marco Gessi3, Giuseppe Maria Vincenzo Barbagallo2. A “polymorphous low-grade neuroepithelial tumor of the young (PLNTY)” diagnosed in an adult. Report of a case and review of the literature. 20-Sep-2021;12:470
How to cite this URL: Giuseppe Broggi1, Francesco Certo2, Roberto Altieri2, Rosario Caltabiano1, Marco Gessi3, Giuseppe Maria Vincenzo Barbagallo2. A “polymorphous low-grade neuroepithelial tumor of the young (PLNTY)” diagnosed in an adult. Report of a case and review of the literature. 20-Sep-2021;12:470. Available from: https://surgicalneurologyint.com/surgicalint-articles/11122/
Date of Submission
19-May-2021
Date of Acceptance
25-Aug-2021
Date of Web Publication
20-Sep-2021
Abstract
Background: Polymorphous low-grade neuroepithelial tumor of the young (PLNTY) is a rare neuropathological entity, recently introduced in neuro-oncology. These tumors, histologically similar to oligodendrogliomas, cause epilepsy, occurring in children and young adults. Only few cases of PLNTY have been described in literature and all reported cases invariably focused on the onset of these tumors in children and young adults.
Case Description: We report the case of a 50-year-old woman suffering from epilepsy since the 1st year of her life. Computed tomography scan and magnetic resonance imaging of the brain documented the presence of a calcified mass involving left temporal lobe. The tumor was surgically excised and the histological examination showed a hypocellular and massively calcified neoplasm with morphological and immunohistochemical features consistent with the diagnosis of “PLNTY.”
Conclusion: A review of the literature revealed that there are 31 cases of PLNTY reported in literature, most of which are children or young adults. The present case represents the second PLNTY diagnosed in a middle-aged adult to the best of our knowledge, suggesting that PLNTY should always be included in the differential diagnosis of low-grade brain tumors, also in adult patients.
Keywords: Adult, Case report, Epilepsy, Neuroepithelial tumor, Polymorphous low-grade neuroepithelial tumor of the young
INTRODUCTION
Low-grade neuroepithelial tumors represent a wide spectrum of brain lesions characterized by heterogeneous histological and molecular features, indolent biological behavior (WHO Grade I and II) and frequent association with early onset of epilepsy in children and young adults.[
We herein report a rare case of PLNTY presenting as an extensively calcified mass located to the left temporal lobe, in a 50-year-old woman with a long time history of drug-resistant epilepsy. A critical review of the scientific literature on PLNTY, highlighting that this tumor may be potentially diagnosed at any age and should not be considered only as a pediatric tumor, is also included.
CASE REPORT
History and clinical findings
A 50-year-old lady, left handed, was referred to the department of neurological surgery, because of a drug-resistant epilepsy. She had a medical history, featured by the onset of seizures since early childhood. The patient was treated over several years with a combination of different anti-epileptic drugs, but she never obtained a complete control of seizures. During the past 2 years before admission, the patient experienced the worsening of crises. These were featured by mental confusion, reduced consciousness, dyskinesia in the upper right limb, and gaze deviation towards the right side. The crises occurred with a frequency of 3–4 episodes per day and occasionally had secondary generalization. She had also hypothyroidism. When the patient was hospitalized, neurological examination revealed moderate mental impairment with slowed speech and gait. Neuroradiological assessment was performed. Brain computed tomography (CT) revealed the presence of a densely calcified lesion involving left temporal lobe and insula [
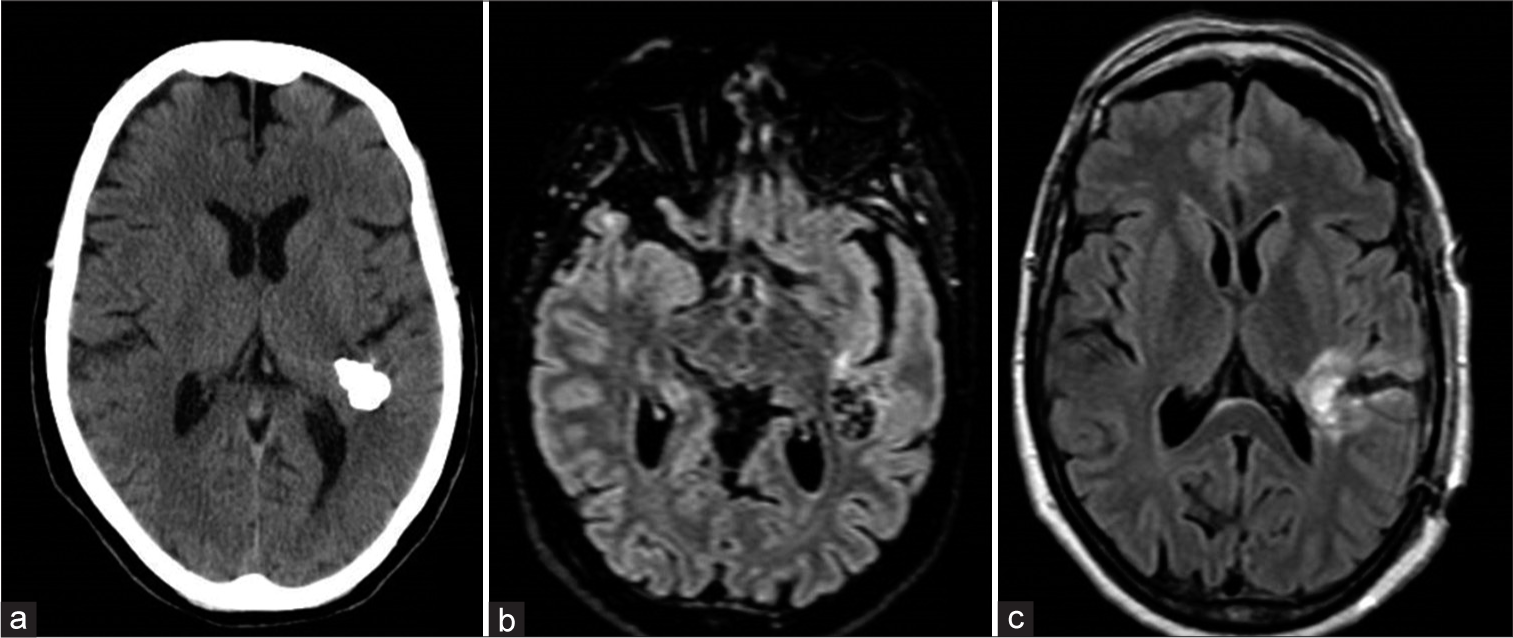
Figure 1:
Preoperative neuroradiological assessment. Computed tomography scan (a) demonstrating the presence of a densely calcified left temporal and insular lesion. FLAIR sequences of magnetic resonance (MR) scan confirmed the presence of the calcified tumor (b). Postoperative MR confirmed the complete resection of the lesion (c).
Surgical procedure
The patient was positioned supine with the left shoulder elevated and head 60° rotated to the right side and fixed by radiolucent three-point Mayfield clamp. Preoperative CT scan with and without iodinate contrast was performed after positioning. Images of postpositioning CT scan were merged with preoperative MRI and used for neuronavigation. Motor evoked potentials, somatosensory evoked potentials and direct monopolar and bipolar cortical and subcortical stimulation were used for neuromonitoring. The intraoperative imaging protocol used in the present case has been previously detailed.[
Postoperative course and outcome
Postoperative MRI confirmed the radical resection of the lesion [
Pathological findings
Grossly, the tumor sample consisted of multiple fragments, markedly hard in consistency, measuring 2 cm in aggregate diameter. Histologic examination showed an extensively calcified and hypocellular lesion [
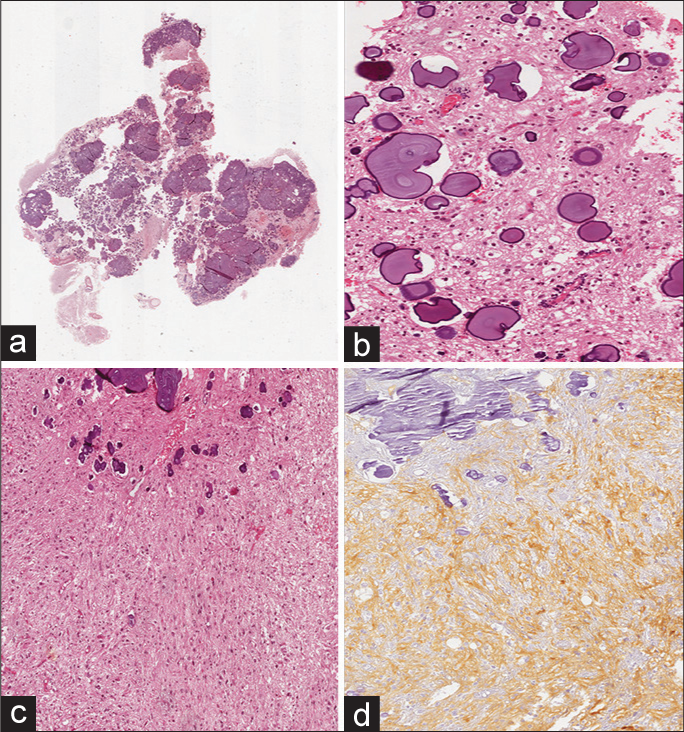
Figure 2:
Histological examination. (a) Low magnification showing the densely calcified nature of the tumor mass (hematoxylin and eosin; original magnification 25×); (b) tumor was mainly composed of small and roundish cells with oligodendroglioma-like morphology intermingled with calcifications ranging from small calcospherules to larger and confluent psammomatous bodies (hematoxylin and eosin; original magnification 150×); (c) a less represented neoplastic component with astrocytic-like morphology was also found (hematoxylin and eosin; original magnification 50×); (d) neoplastic cells were diffusely and strongly stained with CD34 (immunoperoxidase; original magnification 50×).
DISCUSSION
Apart from the most common glial tumors,[
Based on both the facts that PLNTY has a distinct DNA methylation profile and almost invariably presents genetic features involving the MAP kinase pathway components, such as BRAF-V600E mutations or FGFR3- transforming acidic coiled-coil-containing protein 3 (TACC3), FGFR2-KIAA1598, and FGFR2-CTNNA3 fusions, it has now been widely accepted that PLNTY is a distinct tumor entity, belonging to the wide spectrum of low-grade neuroepithelial neoplasms.[
The unusual age of onset of the present case led us to critically perform a literature review and we found that, since its first description,[
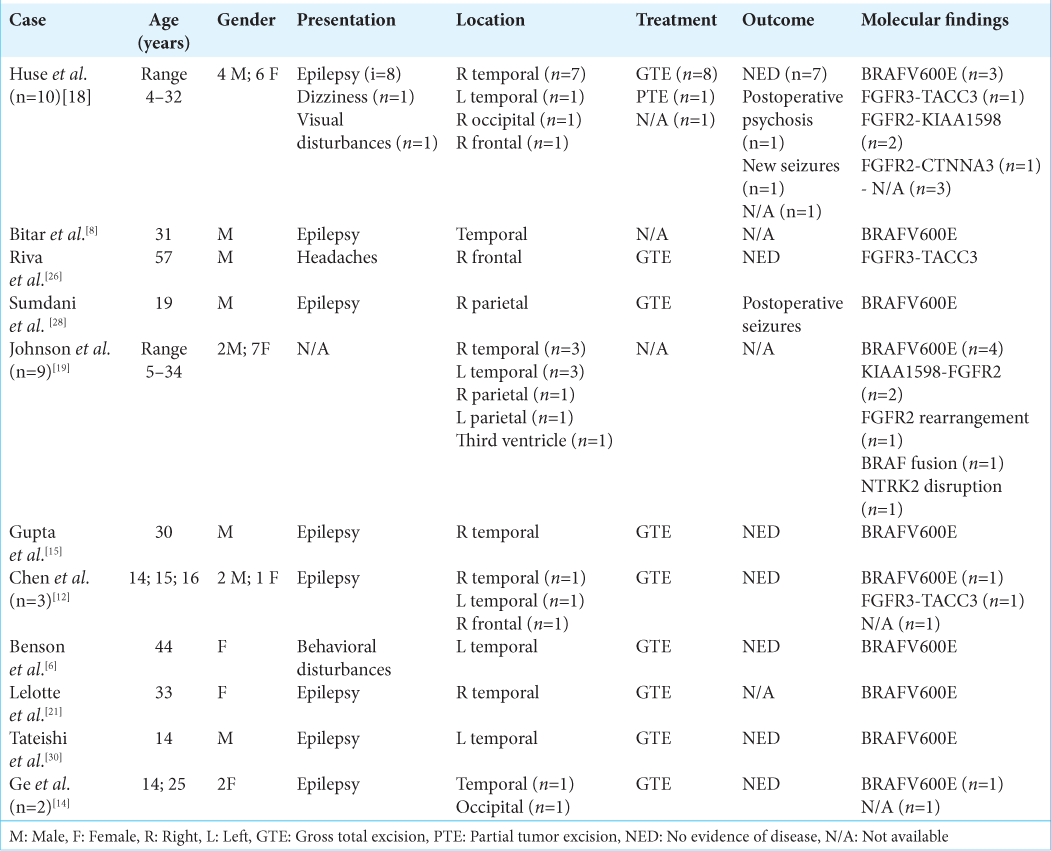
The two most peculiar features of the present case were the age at the diagnosis and the onset as a lowly cellular and diffusely calcified nodule. Even in this regard, we retrospectively reviewed the scientific literature to investigate the presence of papers focusing on the calcified aspect of PLNTY nodules; Johnson et al. in 2019[
On the histopathological point of view, as PLNTY is a round cell with clear perinuclear halo tumor; the differential diagnosis usually includes CNS round/clear cell neoplasms (oligodendroglioma, clear cell ependymoma, and PA with oligodendroglioma-like features) that, however, can be excluded due to the combination of morphological, immunohistochemical (CD34 strong and diffuse expression), and molecular features of PLNTY.[
Moreover, the diagnostic pre- and intra-operative diagnostic assessment used in this case and commonly applied at our institution for all intrinsic brain lesion allowed a thorough study of imaging features of PLNTY. These tumors have common radiological features with calcified cavernous malformations and oligodendrogliomas. i-US revealed a hyperechoic lesion with characteristic posterior shadowing due to the presence of calcium. We also documented that these lesions do not show any 5-ALA induced fluorescence, under ultraviolet light. The present case represents the second confirmed report of PLNTY arising in a middle-aged patient and it led us to reflect on the real need to no longer consider PLNTY as a tumor of the pediatric/young age; although it is likely that, being a slow-growing neoplasm, PLNTY arises at an early age and then is diagnosed late, it is also true that, as PLNTY is associated in almost all cases with the onset of seizures, the hypothesis that a patient can neglect the symptoms and not undergo further diagnostic tests is quite remote.
CONCLUSION
The present case emphasizes the concept that, in the presence of an appropriate clinicoradiological context, neuroradiologists, neurosurgeons, and neuropathologists must include PLNTY in the differential diagnosis even in presence of an adult patient.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Altieri R, Morrone A, Certo F, Parisi G, Buscema G, Broggi G. Tentorial angioleiomyoma: A rare neurosurgical entity. Case report and review of the literature. World Neurosurg. 2019. 130: 506-11
2. Altieri R, Barbagallo D, Certo F, Broggi G, Ragusa M, di Pietro C. Peritumoral microenvironment in high-grade gliomas: From flairectomy to microglia-glioma cross-talk. Brain Sci. 2021. 11: 200
3. Barbagallo D, Caponnetto A, Barbagallo C, Battaglia R, Mirabella F, Brex D. The GAUGAA motif is responsible for the binding between circSMARCA5 and SRSF1 and related downstream effects on glioblastoma multiforme cell migration and angiogenic potential. Int J Mol Sci. 2021. 22: 1678
4. Barbagallo G, Maione M, Peschillo S, Signorelli F, Visocchi M, Sortino G. Intraoperative computed tomography, navigated ultrasound, 5-amino-levulinic acid fluorescence and neuromonitoring in brain tumor surgery: Overtreatment or useful tool combination?. J Neurosurg Sci. 2019. p.
5. Barbagallo GM, Palmucci S, Visocchi M, Paratore S, Attinà G, Sortino G. Portable intraoperative computed tomography scan in image-guided surgery for brain high-grade gliomas: Analysis of technical feasibility and impact on extent of tumor resection. Oper Neurosurg (Hagerstown). 2016. 12: 19-30
6. Benson JC, Summerfield D, Carr C, Cogswell P, Messina S, Gompel JV. Polymorphous low-grade neuroepithelial tumor of the young as a partially calcified intra-axial mass in an adult. AJNR Am J Neuroradiol. 2020. 41: 573-8
7. Berhouma M, Jemel H, Kchir N. Calcified pilocytic astrocytoma of the medulla mimicking a brainstem “stone”. Pathologica. 2008. 100: 408-10
8. Bitar M, Danish SF, Rosenblum MK. A newly diagnosed case of polymorphous low-grade neuroepithelial tumor of the young. Clin Neuropathol. 2018. 37: 178-81
9. Blumcke I, Aronica E, Urbach H, Alexopoulos A, Gonzalez-Martinez JA. A neuropathology-based approach to epilepsy surgery in brain tumors and proposal for a new terminology use for long-term epilepsy-associated brain tumors. Acta Neuropathol. 2014. 128: 39-54
10. Broggi G, Salvatorelli L, Barbagallo D, Certo F, Altieri R, Tirrò E. Diagnostic utility of the immunohistochemical expression of serine and arginine rich splicing factor 1 (SRSF1) in the differential diagnosis of adult gliomas. Cancers (Basel). 2021. 13: 2086
11. Certo F, Altieri R, Maione M, Schonauer C, Sortino G, Fiumanò G. FLAIRectomy in supramarginal resection of glioblastoma correlates with clinical outcome and survival analysis: A prospective, single institution, case series. Oper Neurosurg (Hagerstown). 2021. 20: 151-63
12. Chen Y, Tian T, Guo X, Zhang F, Fan M, Jin H. Polymorphous low-grade neuroepithelial tumor of the young: Case report and review focus on the radiological features and genetic alterations. BMC Neurol. 2020. 20: 123
13. Deb P, Sharma MC, Tripathi M, Chandra PS, Gupta A, Sarkar C. Expression of CD34 as a novel marker for glioneuronal lesions associated with chronic intractable epilepsy. Neuropathol Appl Neurobiol. 2006. 32: 461-8
14. Ge R, Fang HF, Chang YQ, Li Z, Liu CF. Clinicopathological features of polymorphous low-grade neuroepithelial tumor of the young. Zhonghua Bing Li Xue Za Zhi. 2020. 49: 1131-5
15. Gupta VR, Giller C, Kolhe R, Forseen SE, Sharma S. Polymorphous low-grade neuroepithelial tumor of the young: A case report with genomic findings. World Neurosurg. 2019. 132: 347-55
16. Gupta K, Harreld JH, Sabin ND, Qaddoumi I, Kurian K, Ellison DW. Massively calcified low-grade glioma-a rare and distinctive entity. Neuropathol Appl Neurobiol. 2014. 40: 221-4
17. Hewer E, Knecht U, Ulrich CT. Two adult cases of massively calcified low-grade glioma: Expanding clinical spectrum of an emerging entity. Neuropathology. 2016. 36: 508-9
18. Huse JT, Snuderl M, Jones DT, Brathwaite CD, Altman N, Lavi E. Polymorphous low-grade neuroepithelial tumor of the young (PLNTY): An epileptogenic neoplasm with oligodendroglioma-like components, aberrant CD34 expression, and genetic alterations involving the MAP kinase pathway. Acta Neuropathol. 2017. 133: 417-29
19. Johnson DR, Giannini C, Jenkins RB, Kim DK, Kaufmann TJ. Plenty of calcification: Imaging characterization of polymorphous low-grade neuroepithelial tumor of the young. Neuroradiology. 2019. 61: 1327-32
20. Kim YE, Shin HJ, Suh YL. Pilocytic astrocytoma with extensive psammomatous calcification in the lateral ventricle: A case report. Childs Nerv Syst. 2012. 28: 649-52
21. Lelotte J, Duprez T, Raftopoulos C, Michotte A. Polymorphous low-grade neuroepithelial tumor of the young: Case report of a newly described histopathological entity. Acta Neurol Belg. 2020. 120: 729-32
22. Louis DN, Ohgaki H, Weistler OD, Cavenee WK.editors. WHO Classification of Tumours of the Central Nervous System. Lyon: International Agency for Research on Cancer (IARC); 2016. p.
23. Nagaishi M, Arai M, Osawa T, Yokoo H, Hirato J, Yoshimoto Y. An immunohistochemical finding in glioneuronal lesions associated with epilepsy: The appearance of nestin-positive, CD34-positive and tau-accumulating cells. Neuropathology. 2011. 31: 468-75
24. Niimi M, Yoshida K, Mayanagi K, Kawase T. Extensive and dense calcification in the core of a ventrally exophytic brainstem glioma. Brain Tumor Pathol. 2002. 19: 101-3
25. Prabowo AS, Iyer AM, Veersema TJ, Anink JJ, Schoutenvan Meeteren AY, Spliet WG. BRAF V600E mutation is associated with mTOR signaling activation in glioneuronal tumors. Brain Pathol. 2014. 24: 52-66
26. Riva G, Cima L, Villanova M, Ghimenton C, Sina S, Riccioni L. Low-grade neuroepithelial tumor: Unusual presentation in an adult without history of seizures. Neuropathology. 2018. 38: 557-60
27. Sloan EA, Chiang J, Villanueva-Meyer JE, Alexandrescu S, Eschbacher JM, Wang W. Intracranial mesenchymal tumor with FET-CREB fusion-a unifying diagnosis for the spectrum of intracranial myxoid mesenchymal tumors and angiomatoid fibrous histiocytoma-like neoplasms. Brain Pathol. 2020. 31: e12918
28. Sumdani H, Shahbuddin Z, Harper G, Hamilton L. Case report of rarely described polymorphous low-grade neuroepithelial tumor of the young and comparison with oligodendroglioma. World Neurosurg. 2019. 127: 47-51
29. Tamura M, Kohga H, Ono N, Zama A, Shibasaki T, Horikoshi S. Calcified astrocytoma of the amygdalo-hippocampal region in children. Childs Nerv Syst. 1995. 11: 141-4
30. Tateishi K, Ikegaya N, Udaka N, Sasame J, Hayashi T, Miyake Y. BRAF V600E mutation mediates FDG-methionine uptake mismatch in polymorphous low-grade neuroepithelial tumor of the young. Acta Neuropathol Commun. 2020. 8: 139

