

- Department of Neurosurgery, Dhaka Medical College Hospital, Dhaka, Bangladesh.
- Medical Student, Dhaka Medical College Hospital, Dhaka, Bangladesh.
Correspondence Address:
Mobin Ibne Mokbul, Medical Student, Dhaka Medical College Hospital, Dhaka, Bangladesh.
DOI:10.25259/SNI_325_2023
Copyright: © 2023 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Abdullah Al-Noman1, Mobin Ibne Mokbul2, Nadia Hossain2, Md. Sumon Rana1, Md. Motasimul Hasan1, Md. Shafiqul Islam1. A young female of Cowden syndrome presenting with Lhermitte-Duclos disease: An illustrative case. 25-Aug-2023;14:296
How to cite this URL: Abdullah Al-Noman1, Mobin Ibne Mokbul2, Nadia Hossain2, Md. Sumon Rana1, Md. Motasimul Hasan1, Md. Shafiqul Islam1. A young female of Cowden syndrome presenting with Lhermitte-Duclos disease: An illustrative case. 25-Aug-2023;14:296. Available from: https://surgicalneurologyint.com/surgicalint-articles/12517/
Date of Submission
11-Apr-2023
Date of Acceptance
22-Jul-2023
Date of Web Publication
25-Aug-2023
Abstract
Background: Lhermitte-Duclos Disease (LDD), or dysplastic gangliocytoma, which is a benign hamartomatous condition involving the cerebellum, has a possible association with Cowden syndrome (CS), a rare autosomal dominant disorder due to germline mutations in the phosphatase and tensin homolog (PTEN) tumor-suppressor gene in chromosome 10. Combined CS and LDD cases are rarely reported in the literature.
Case Description: We present here a case of a young female patient presented at the emergency department with a severe headache associated with vertigo, vomiting, and cerebellar ataxia. A magnetic resonance imaging scan revealed mixed intensity posterior fossa lesion with almost preserved cerebellar cortical striations. Her facial skin had extensive trichilemmoma. Her symptoms improved after the excision of the posterior fossa lesion through suboccipital craniotomy and histopathology revealed LDD.
Conclusion: In a low-resource country where genetic testing for neurosurgical condition is still inadequate, we used the validated Cleveland Clinic Adult Clinical Scoring for PTEN Testing and the patient had an 82–98% chance for a PTEN gene mutation. Finally, she along with her family was adequately counseled and was advised for regular screening and monitoring since it is a premalignant condition where early detection is imperative if any cancer arises in the near future and is now under our follow-up.
Keywords: Cerebellar ataxia, Cowden syndrome, Lhermitte-Duclos disease, Malignancy, Phosphatase and tensin homolog, PTEN
INTRODUCTION
Multiple hamartoma syndrome eponym as Cowden syndrome (CS) is a rare autosomal dominant disorder.[
In addition, LDD is a rare disorder characterized by slowly progressive unilateral dysplastic gangliocytoma of the cerebellar cortex first recognized in 1920 by Lhermitte and Duclos.[
Adult LDD is considered as one of the pathognomonic criteria for diagnosing CS, along with mucocutaneous lesions, facial trichilemmomas, acral keratoses, and papillomatous papules.[
Clinical symptoms generally arise from/are related to mass effect changes in the posterior fossa and can include headache, nausea, and visual disturbances and sometimes, might cause blindness too. These early symptoms can be controlled by sometimes through shunt placement (e.g., Ventriculoperitoneal shunting) which can help with the release of cerebrospinal fluid blocking. The severity of symptoms can vary depending on the size of the lesion, and patients with isolated LDD may be asymptomatic for years. If the lesion grows large enough, patients may also exhibit signs of cerebellar dysfunction and obstructive hydrocephalus.[

CASE DESCRIPTION
Patient presentation
A young female patient (27 years old) presented at the Emergency Department of Dhaka Medical College Hospital on November 2020 with a severe headache associated with vertigo and vomiting. The neurological assessment revealed average intelligence and marked cerebellar ataxia. Papilledema was noted on fundoscopy. She had 2 years of history of headache with nausea. After primary resuscitation, detailed history and examination was carried out and previous records were checked. Clinical examination revealed mild anemia and firm flat-topped brownish papules over the face and hyperkeratosis over several areas in the body. Multiple lipomas were palpated in the limbs and torso. She also had neck swelling. Physical examination findings are shown in
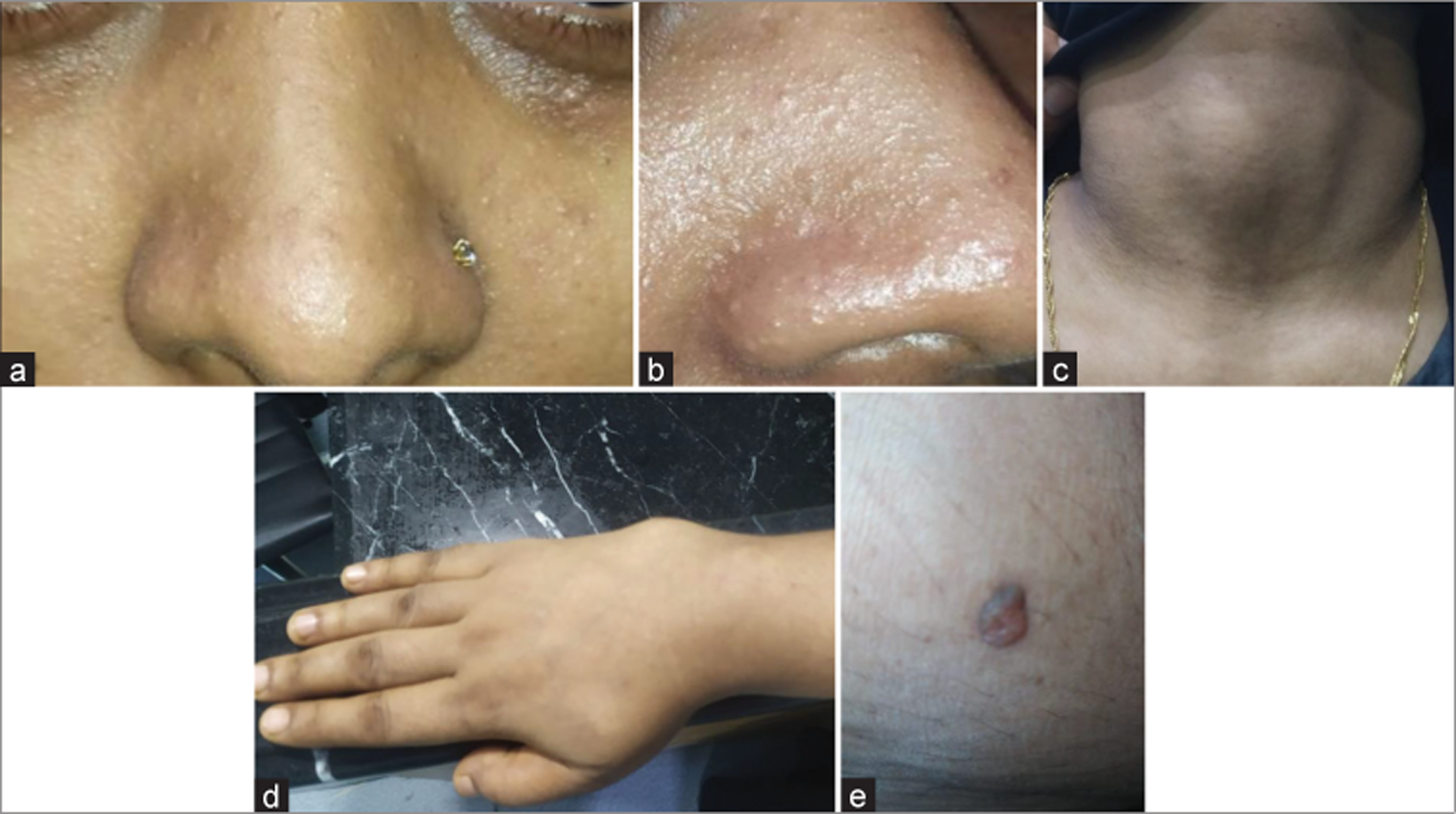
Figure 1:
(a) Photo of face showing trichilemmoma (anterior view). (b) Photo of face showing trichilemmoma (anterolateral view). (c) Patient had neck swelling. Later confirmed by ultrasonogram as multinodular goiter with thyroiditis. (d) Soft swelling in lateral aspect of wrist joint. (e) Trichilemmoma of the thigh.
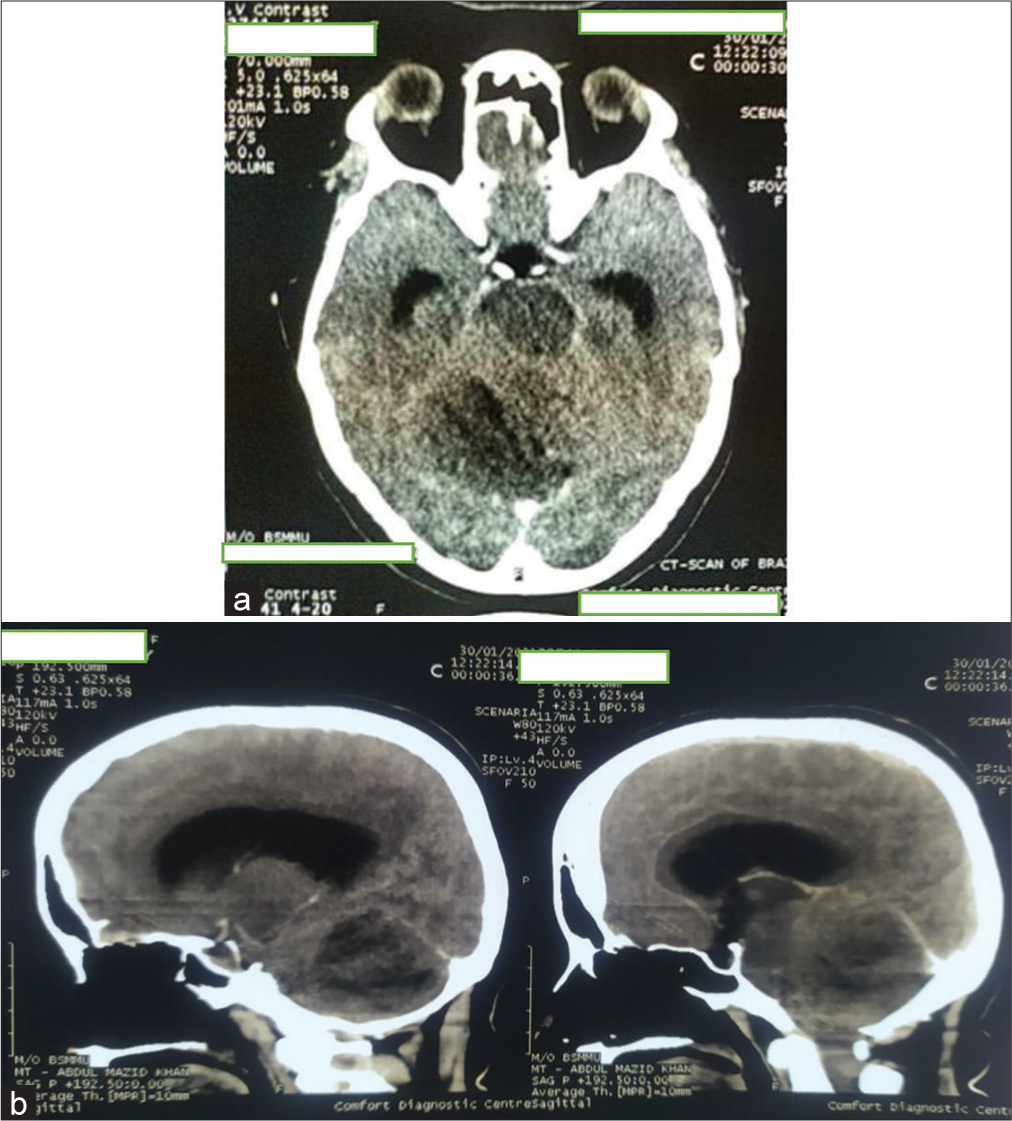
Figure 2:
Initial contrast-enhanced computed tomography (CT) scan of brain (a) axial image and (b) sagittal section showing ill-defined hypodense area with mild heterogeneous enhancement in right cerebellum involving middle cerebellar peduncle having perilesional edema and mass effect compressing pons, effacement of 4th ventricle resulting mild dilatation of both lateral and 3rd ventricles.
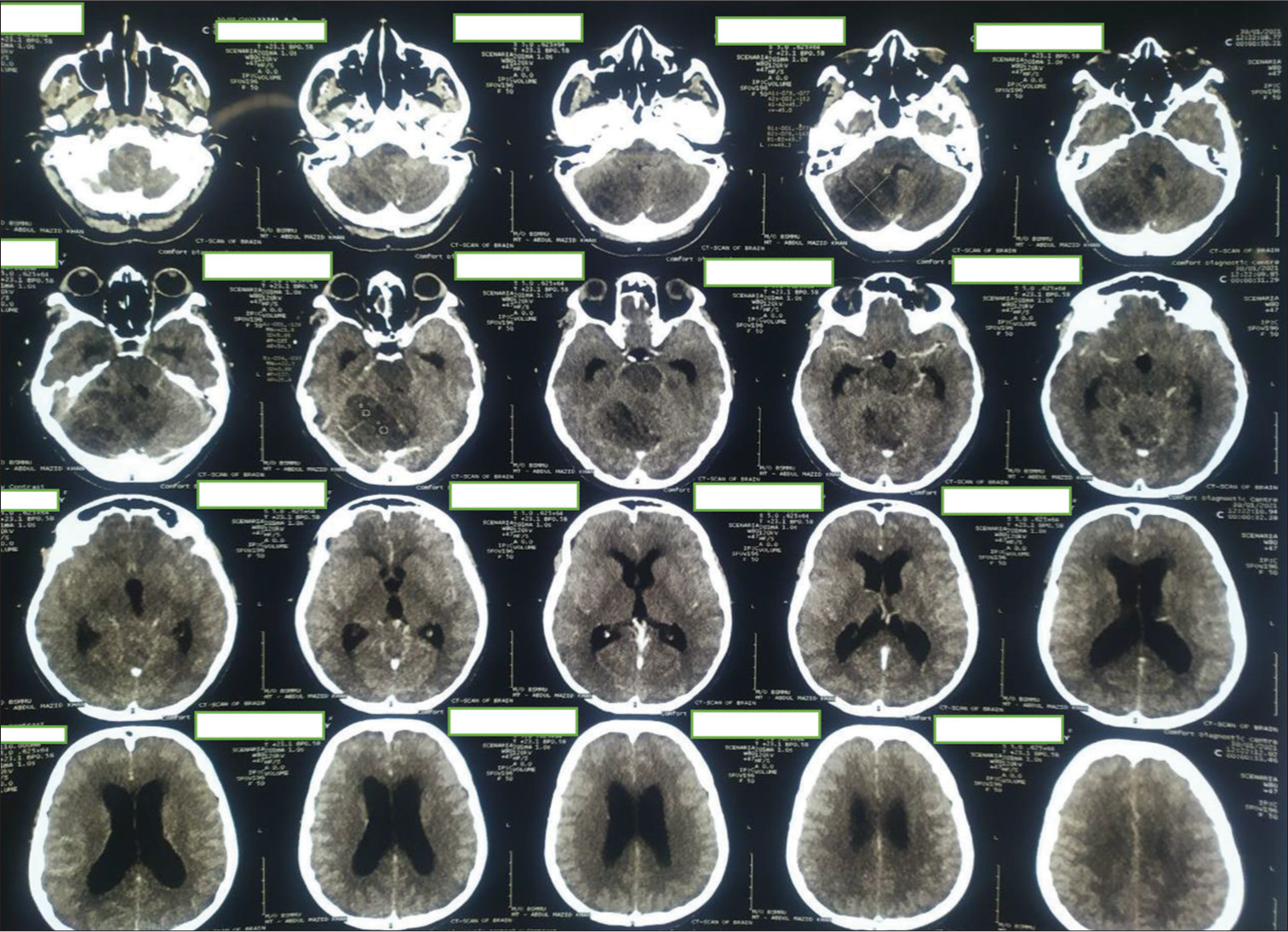
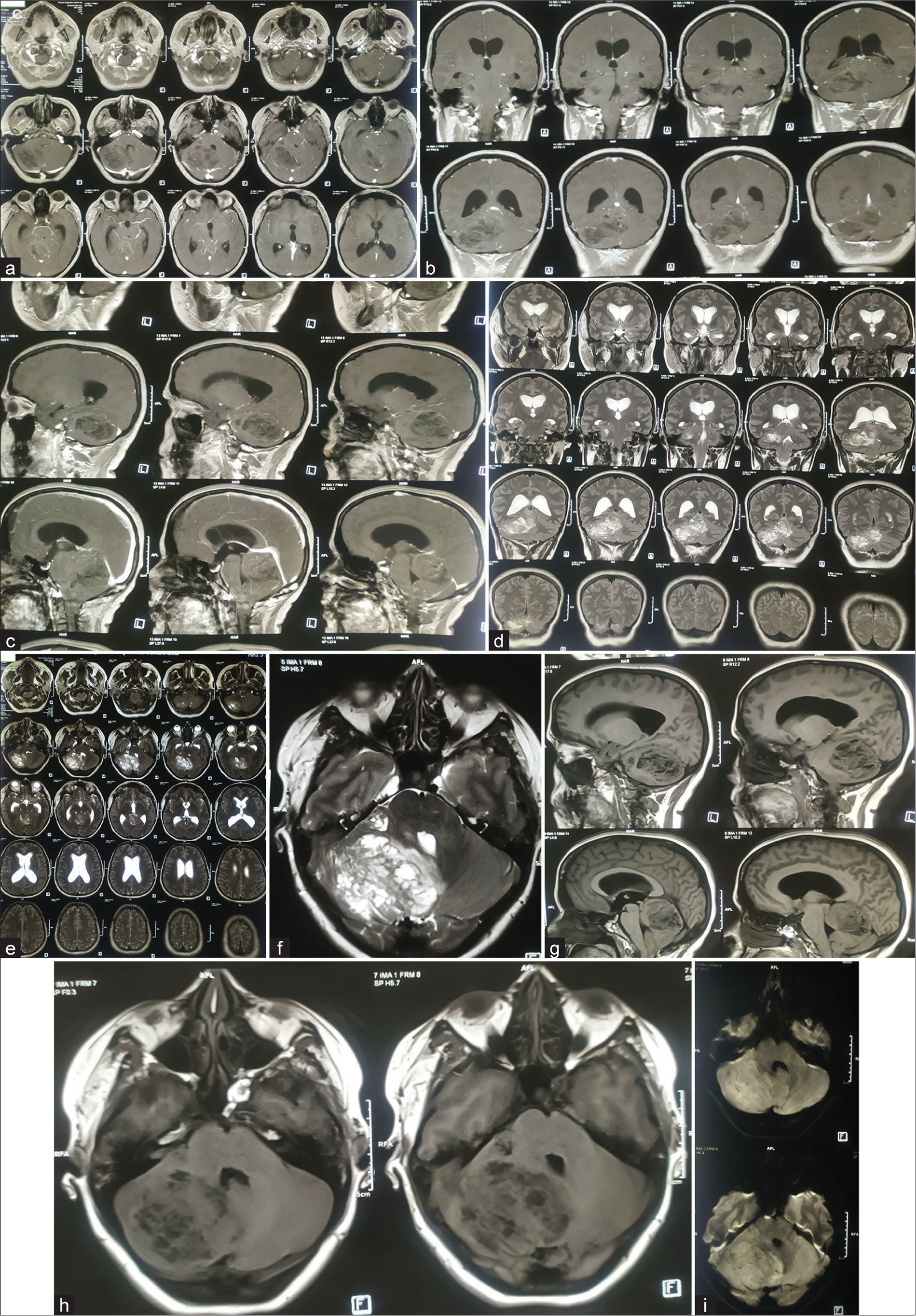
Figure 4:
(a) Preoperative magnetic resonance imaging (MRI) of the brain of the patient. (a) T1 contrast axial sequences, (b) T1 contrast: coronal sequence, (c) T1 contrast: sagittal sequence, (d) T2-weighted image (T2WI) coronal Sequence, (e) T2WI axial sequence, (f) T2WI axial image focusing on cerebellar lesion, (g) T1-weighted image (T1WI) sagittal sequence, (h) T1 WI axial, and (i) diffusion-weighted imaging sequence. Contrast-enhanced MRI scan of brain showing mild heterogeneously enhancing, tigroid, or striated mass lesion on right cerebellum involving middle cerebellar peduncle having perilesional edema and mass effect compressing pons, effacement of 4th ventricle resulting tri-ventriculomegaly; suggestive of low-grade glioma. Differential – medulloblastoma, Lhermitte-Duclos Disease (LDD) (which was later confirmed in histopathology to be LDD).
The patient also gave us a recent history of cholecystectomy due to cholelithiasis. Before making the diagnosis of cholelithiasis, an ultrasonogram (USG) was done due to her complain of frequent abdominal pain [
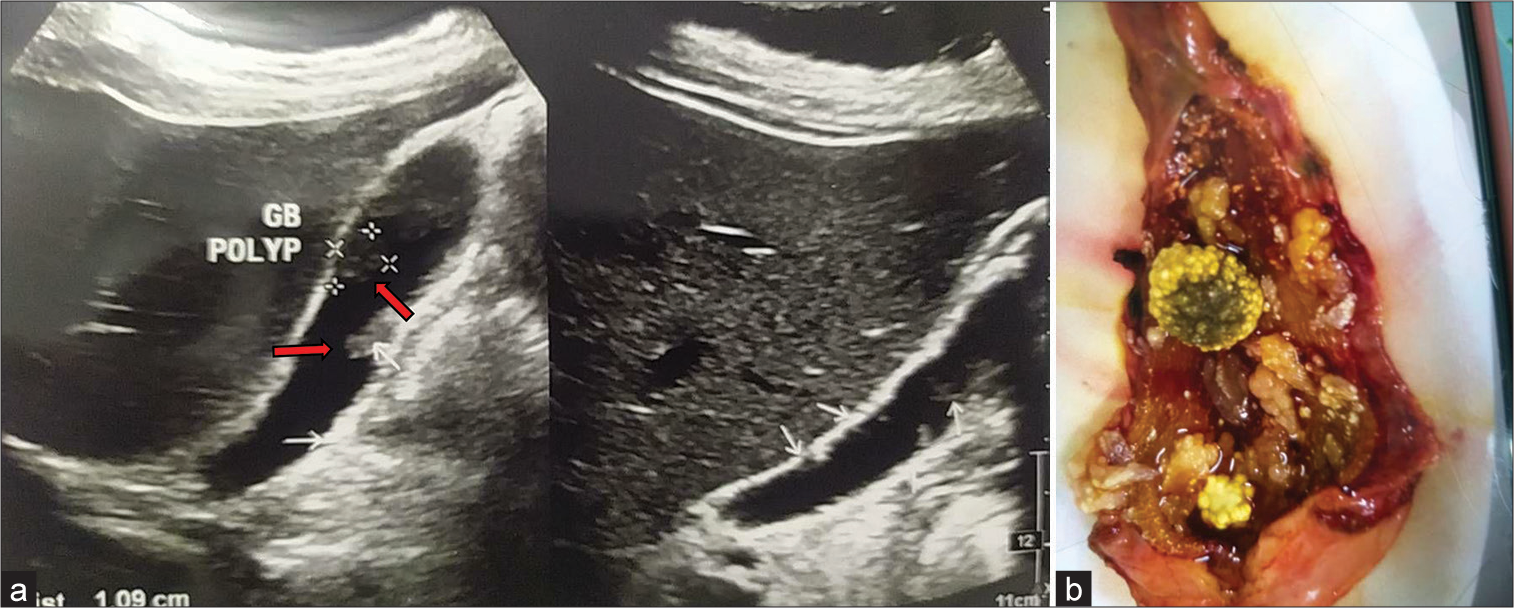
Figure 5:
(a) Multiple gallbladder (GB) polyps with cholelithiasis; two mobile bright echogenic structures (marked in red arrows) in the lumen of gallbladder casting distal acoustic shadows (Larger one 1.5 × 0.6 cm). Numerous soft-tissue echogenic structures without acoustic shadows are seen adherent with both anterior and posterior wall. (b) Multiple GB polyps (5 or more) with pigmented stones after cholecystectomy.
Management
The patient was monitored closely to figure out the treatment plan. With decisions from a multidisciplinary medical board, our patient underwent surgical excision of the posterior fossa lesion through suboccipital craniotomy which resulted in improvement of cerebellar symptoms. The patient underwent total resection of the cerebellar lesion.
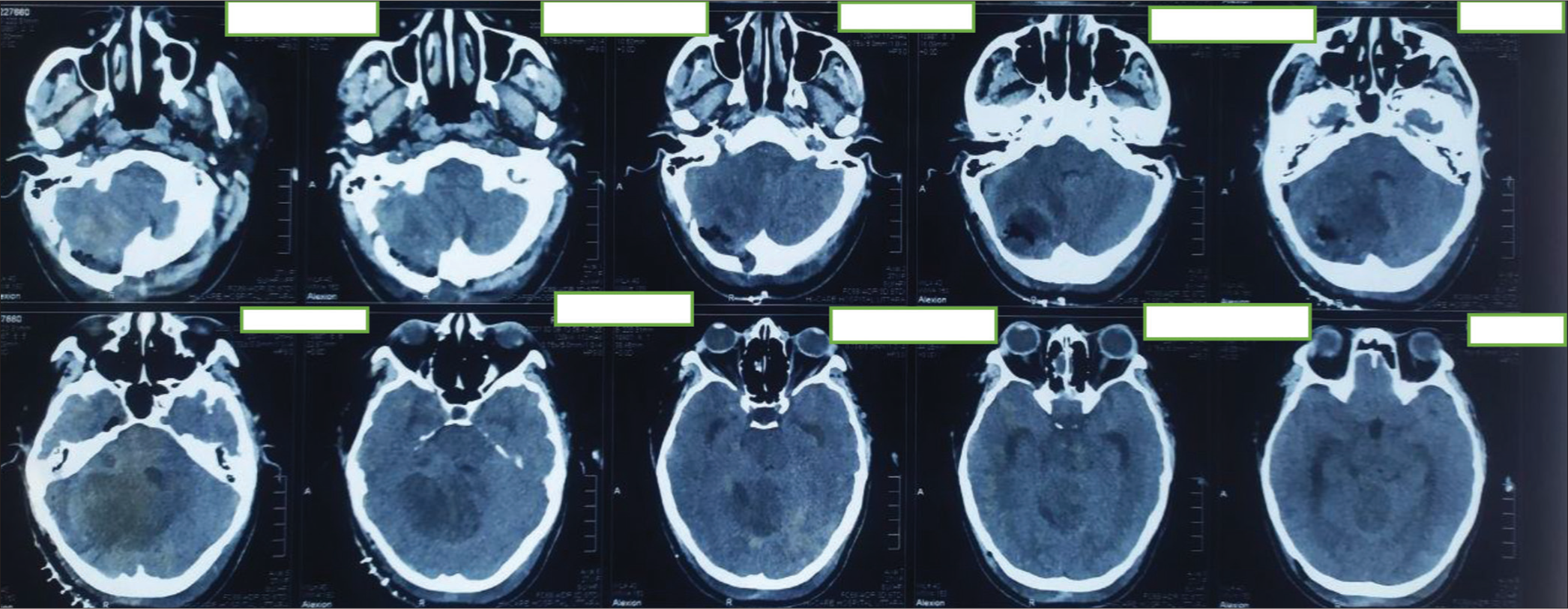
Figure 7:
First postoperative day computed tomography scan. Large irregular mixed density area seen in the right cerebellum and extends in the right cerebellopontine angle region. Mass effect is evident by effacement of 4th ventricle and shifting of mid line towards left right side. Postoperative pneumocephalus and edema seen in right cerebellum and temporal lobe.

Figure 8:
Histopathology report of the cerebellar SOL suggestive of Lhermitte-Duclos Disease. These reveal glial tissue containing elongated fibers and small vacuolated spaces. It also reveals scattered large ganglion cells with irregular margin and large nuclei. Normal cerebellar tissue is not present in the specimen.
DISCUSSION
Our previous provisional diagnosis was low-grade glioma or medulloblastoma from the initial radiological imaging at presentation. However, due to all these physical examination findings, clinical history, imaging, and histopathology, we concluded this case to be an LDD with CS. The diagnostic criteria for CS are as follows in

While significant functional research has been conducted, the full function of PTEN, the phosphatase enzyme is yet unknown.[
Genetic testing is confirmatory for CS diagnosis that typically involves identifying mutations or variants in the PTEN gene. In developed nations, there are certain standardized genetic assessments to diagnose CS such as multiplex ligation-dependent probe amplification, traditional Sanger sequencing, and next-generation sequencing (NGS). multiplex ligation-dependent probe amplification (MLPA®), a technique used to identify large deletions or duplications in the PTEN gene, involves hybridizing specific probes to the deoxyribonucleic acid (DNA) and subsequently amplifying them to detect copy number changes.[
That is why we used Cleveland Clinic Adult Clinical Scoring for PTEN Testing (can be accessed here:

CONCLUSION
It is captivating how a single genetic mutation can lead to a broad variety of abnormalities, as seen in our case, necessitating surgery, family counseling, and monitoring the patient carefully. While genetic testing for such disorders is not possible in low-resource country, thorough clinical examination and proper laboratory investigations and imaging should be done as early as possible to lessen the complications and detect or exclude associated malignancies that might appear someday in the future. Proper counseling for early identification of precancerous symptoms and regular follow-up is instrumental in treating this rare genetic condition.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Publication of this article was made possible by the James I. and Carolyn R. Ausman Educational Foundation.
Conflicts of interest
There are no conflicts of interest.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
1. Barkovich AJ, editors. Pediatric Neuroimaging. Philadelphia, PA: Lippincott Williams and Wilkins; 2005. p.
2. Bubien V, Bonnet F, Brouste V, Hoppe S, Barouk-Simonet E, David A. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet. 2013. 50: 255-63
3. Casperson BK, Anaya-Baez V, Kirzinger SS, Sattenberg R, Heidenreich JO. Coexisting MS and Lhermitte-Duclos disease. J Radiol Case Rep. 2010. 4: 1-6
4. Daly MB, Pal T, Berry MP, Buys SS, Dickson P, Domchek SM. Genetic/familial high-risk assessment: Breast, ovarian, and pancreatic, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2021. 19: 77-102
5. Farooq A, Walker LJ, Bowling J, Audisio RA. Cowden syndrome. Cancer Treat Rev. 2010. 36: 577-83
6. Gama I, Almeida L. Lhermitte-Duclos disease associated to Cowden syndrome: De novo diagnosis and management of these extremely rare syndromes in a patient. BMJ Case Rep. 2017. 2017: bcr2016217974
7. Giorgianni A, Pellegrino C, De Benedictis A, Mercuri A, Baruzzi F, Minotto R. Lhermitte-Duclos disease. A case report. Neuroradiol J. 2013. 26: 655-60
8. Gustafson S, Zbuk KM, Scacheri C, Eng C. Cowden syndrome. Semin Oncol. 2007. 34: 428-34
9. Joo G, Doumanian J. Radiographic findings of dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) in a woman with Cowden syndrome: A case study and literature review. J Radiol Case Rep. 2020. 14: 1-6
10. Kulkantrakorn K, Awwad EE, Levy B, Selhorst JB, Cole HO, Leake D. MRI in Lhermitte-Duclos disease. Neurology. 1997. 48: 725-31
11. Lhermitte J, Duclos P. On a diffuse ganglioneuroma of the cerebellar cortex. Bull Assoc Fr Etud Cancer. 1920. 9: 99-107
12. Masmoudi A, Chermi ZM, Marrekchi S, Raida BS, Boudaya S, Mseddi M. Cowden syndrome. J Dermatol Case Rep. 2011. 5: 8-13
13. Nowak DA, Trost HA. Lhermitte-Duclos disease (dysplastic cerebellar gangliocytoma): A malformation, hamartoma or neoplasm?. Acta Neurol Scand. 2002. 105: 137-45
14. Pritchard CC, Smith C, Marushchak T, Koehler K, Holmes H, Raskind W. A mosaic PTEN mutation causing Cowden syndrome identified by deep sequencing. Genet Med. 2013. 15: 1004-7
15. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977. 74: 5463-7
16. Tan MH, Mester J, Peterson C, Yang Y, Chen JL, Rybicki LA. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet. 2011. 88: 42-56
17. Yehia L, Eng C, Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW, editors. PTEN Hamartoma tumor syndrome. GeneReviews®. Seattle, WA: University of Washington, Seattle; 2001. p. 1993-2023, Available from: https://www.ncbi.nlm.nih.gov/books/NBK1488 [Last accessed on 2021 Feb 11]
18. Yehia L, Ngeow J, Eng C. PTEN-opathies: From biological insights to evidence-based precision medicine. J Clin Invest. 2019. 129: 452-64

