

Adult gangliocytoma arising within the lateral ventricle: A case report and review of the literature
- Department of Surgery, Section of Neurosurgery, Max Rady College of Medicine, University of Manitoba, Canada.
- Department of Pathology, Health Sciences Centre and University of Manitoba, Winnipeg, Manitoba, Canada.
Correspondence Address:
Dr. Jason Beiko, Section of Neurosurgery, Department of Surgery, Max Rady College of Medicine, University of Manitoba, Winnipeg, Manitoba, Canada.
DOI:10.25259/SNI_814_2021
Copyright: © 2022 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Norah Alarifi1, Marc R. Del Bigio2, Jason Beiko1. Adult gangliocytoma arising within the lateral ventricle: A case report and review of the literature. 12-Jan-2022;13:11
How to cite this URL: Norah Alarifi1, Marc R. Del Bigio2, Jason Beiko1. Adult gangliocytoma arising within the lateral ventricle: A case report and review of the literature. 12-Jan-2022;13:11. Available from: https://surgicalneurologyint.com/surgicalint-articles/11341/
Date of Submission
14-Aug-2021
Date of Acceptance
14-Dec-2021
Date of Web Publication
12-Jan-2022
Abstract
Background: Gangliocytomas are rare neuronal tumors with an incidence of
Case Description: We report a case of an intracranial gangliocytoma arising within the lateral ventricle in a 66-year-old female. Magnetic resonance imaging of the brain showed a diffusely enhancing lobulated mass situated within the frontal horn of the right lateral ventricle with extension into the foramen of Monro and obstructive hydrocephalus. The patient underwent an interhemispheric transcallosal approach with gross total resection and relief of her hydrocephalus. Pathological examination showed clusters of highly pleomorphic neuron-like cells without evidence of neoplastic glial cells. Histopathological and immunohistochemistry findings were consistent with the diagnosis of gangliocytoma (World Health Organization Grade 1).
Conclusion: Gangliocytomas are rare low-grade CNS neoplasms that can present in an older population within unusual locations and should be included within the differential whenever a suspicious lesion is encountered.
Keywords: Adult, Central nervous system, Gangliocytoma, Ganglioglioma, Ventricle
INTRODUCTION
Gangliocytomas are rare central nervous system (CNS) neoplasms that consist of mature, large neuron-like cells.[
CASE REPORT
Clinical presentation
A 66-year-old female with a medical history of hypertension and depression presented with a 10-month history of progressive generalized weakness, fatigability, mild nonspecific headaches, and penultimately confusion. Her symptoms significantly worsened the week before her presentation, suffering multiple falls as a result. In the emergency department, she did not endorse any visual changes, seizures or seizure-like activity, any specific pattern to her headaches, nausea or vomiting, or any other constitutional symptoms. Her neurological examination was within the normal limits with no apparent focal neurological deficits.
Computed tomography scan of the head showed an intraventricular mass lesion with associated obstructive hydrocephalus [
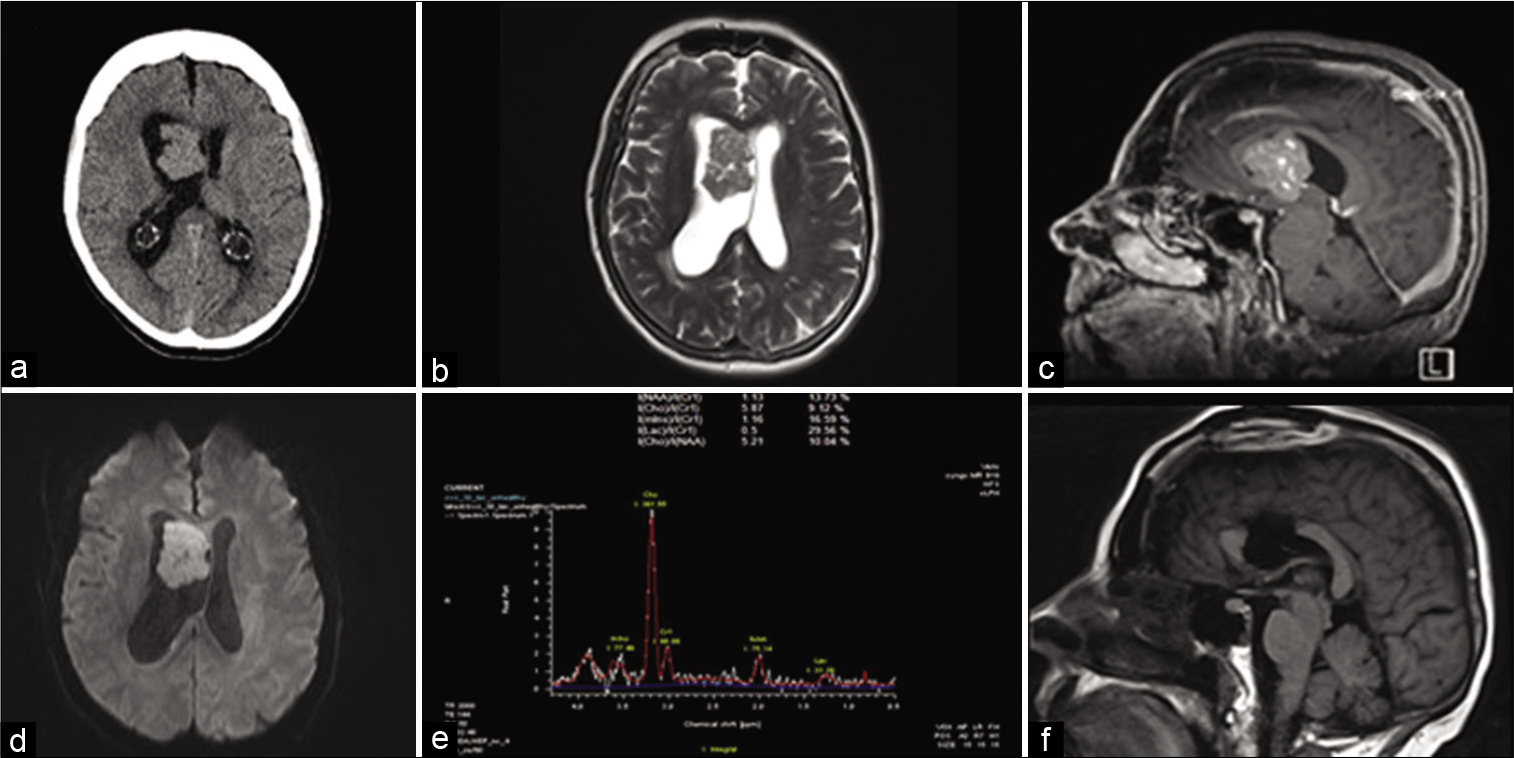
Figure 1:
Radiological features. (a) CT head showing right frontal lesion with ventriculomegaly. (b) Axial T2-weighted MRI showing lesion with isointense signal. (c) Sagittal T1 + Gadolinium showing diffuse enhancement of the lesion. (d) Diffusion-weighted image (DWI) showing mild diffusion restriction within the lesion. (e) MR spectroscopy showing markedly elevated choline peak and diminished NAA peak. (f) Postoperative sagittal T1-weighted MRI showing gross total resection.
Surgical intervention
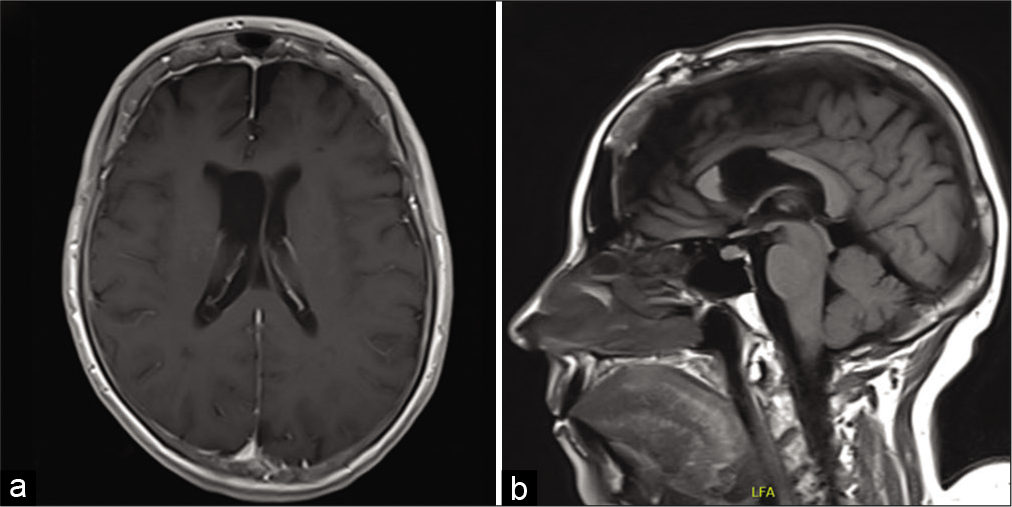
Shortly after presentation, she underwent a craniotomy; interhemispheric dissection and anterior callosotomy allowed entry into the lateral ventricle. The tumor was immediately identifiable by its tan color and lobulated appearance. It had two consistencies with one part being easily aspirated and a second firmer part that required ultrasonic aspiration. The inside of the tumor contained numerous blood vessels with pronounced veins. Of note, hemostasis was easily achievable with electrocautery. Anteriorly and medially, it had some obvious attachments that required microsurgical dissection primarily from the septum pellucidum. Eventually, the dissection led down to the ipsilateral foramen of Monro and into the third ventricle. Once the tumor was freed from its attachment points, complete removal was achieved. The patient recovered well from her surgery and was neurologically intact without any focal deficits. Postoperative MRI showed gross total resection of the lesion with improving ventriculomegaly [
Pathology
Intraoperative evaluation of smeared and frozen samples showed a pleomorphic neoplasm, without clear high-grade or papillary features. The remaining tumor specimens were fixed in formalin and embedded in paraffin. Sections were stained for hematoxylin and eosin, and a broad panel of immunohistochemical stains to determine glial and neuronal lineage. Histologically the superficial aspects of the tumor had a smooth surface [
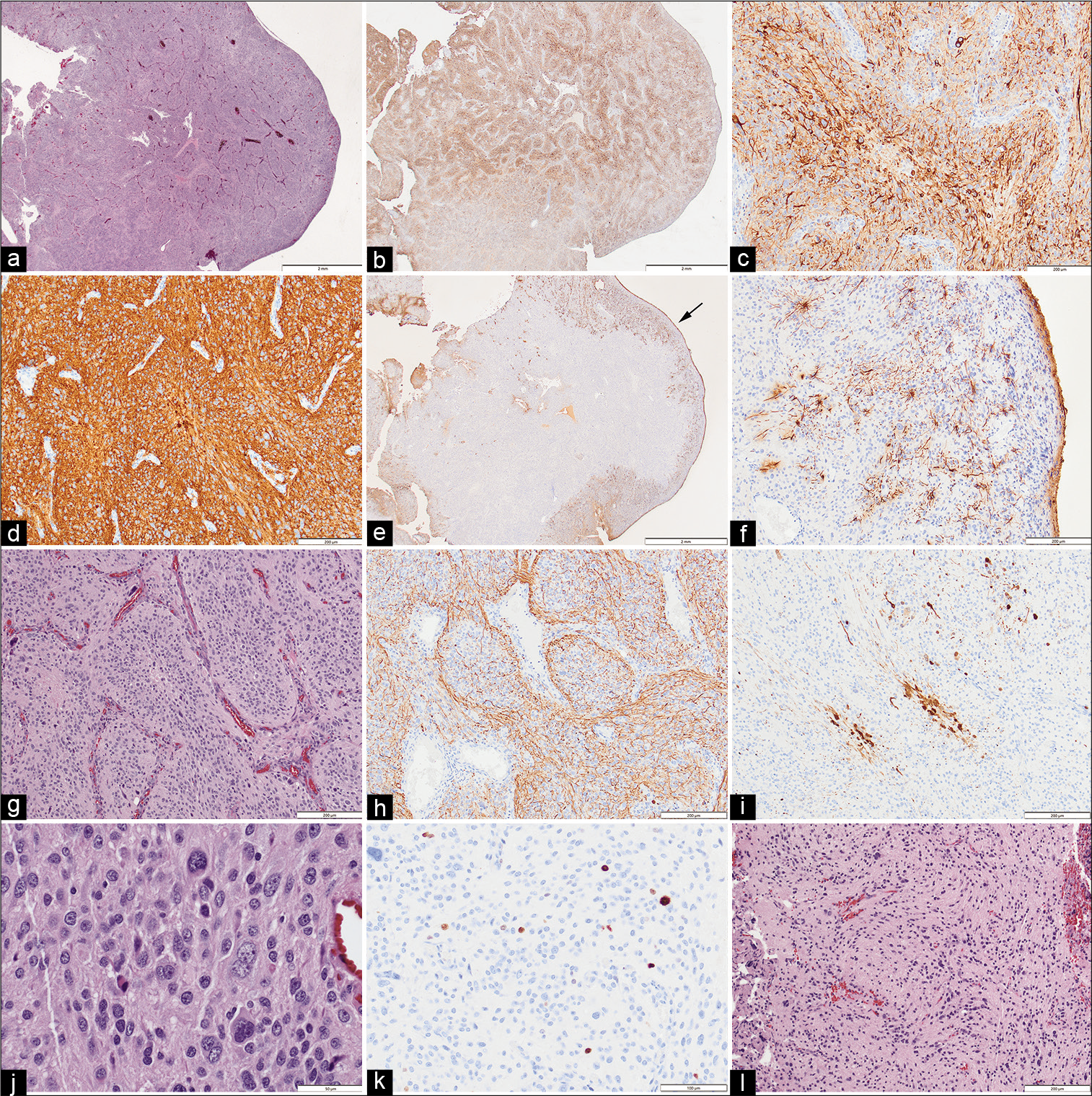
Figure 3:
(a) The superficial aspects of the tumor adjacent to the lateral ventricle had a smooth surface (Hematoxylin & Eosin). (b) Immunoreactivity for nonphosphorylated neurofilament (npNF) was present throughout the tumor and at low magnification had a variegated appearance. (c) The cells of the lobules, which were of varied size, were uniformly immunoreactive for npNF. (d) Tumor cells were uniformly immunoreactive for synaptophysin (as well as CD56 and Pgp9.5, not shown). (e) Immunoreactivity for glial fibrillary acidic protein (GFAP) was restricted to the tumor surface (arrow). (f) GFAP immunoreactive glial cells had non-neoplastic stellate appearance and were restricted to the tumor surface. (g) Intermediate magnification of the tumor core showed cellular lobules outlined by narrow vascular septae (H&E). (h) Neurofilament immunostain showed axon-like processes mainly at the periphery of the lobules. (i) In some lobule cores, the neuron-like cells were also immunoreactive for calbindin 2 (calretinin). (j) High magnification shows that the majority of the tumor cells are of medium to large size, often with neuron-like appearance. (k) Scattered nuclei were immunoreactive for Ki67 (extremely rare mitotic figures were identified, not shown). (l) Rare fragments of the tumor in the CUSA sample, presumed to represent the attachment site, had a more glial appearance with scattered atypical neurons (H&E). Original magnifications - (a, b, e) x12.5; (c, d, f-i, l) x100; (j) x400; (k) x200
DISCUSSION
Gangliocytomas were described in the first edition of the WHO classification of nervous system tumors in 1979.[
Ganglion cell tumors have a controversial nosology. They are regarded as a spectrum of neoplasms predominated by neuronal cells. Gangliogliomas and gangliocytomas occupy opposite ends of the spectrum, mainly based on the presence of neoplastic glial cells.[
Their histogenesis remains elusive. Some have been observed to have common cytological abnormalities with certain neuronal migration disorders, such as hemimegalencephaly.[
Epidemiology
Ganglion cell tumors are rare, accounting for 1–3% of CNS tumors with gangliogliomas more common than gangliocytomas.[
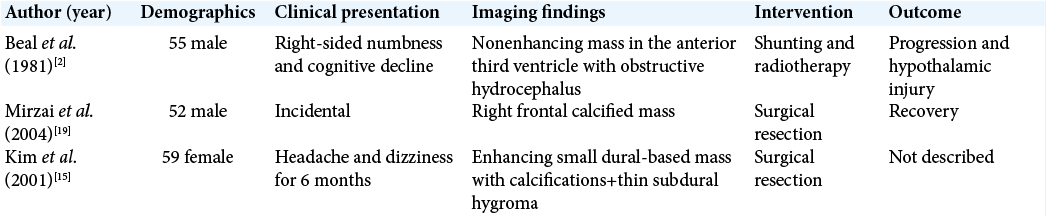
CNS location and clinical presentation
Ganglion cell tumors are reported throughout the CNS. Gangliocytomas specifically are reported to arise within the cerebral hemispheres, temporal lobe especially, and the spinal cord.[
Seizures are a common presentation of CNS gangliocytomas.[
Radiological features
Gangliocytomas can resemble gangliogliomas in their imaging features.[
Our patient’s MRI findings [
Pathology
Ganglion cell tumors usually appear as well-circumscribed, firm, nodular, and frequently calcified lesions with some having associated cysts.[
The lesion in our case exhibited features of a glioneuronal neoplasm with its ganglionic cell elements. The tumor core is purely neuronal, with demonstrated positivity for synaptophysin, CD56, nonphosphorylated neurofilament, Pgp9.5, NF, and NeuN as to be expected of a gangliocytoma. Furthermore, the core lacked a glial component, and the GFAP-positive cells were confined to the tumor attachment and the surfaces exposed to the ventricles, which would be atypical of ganglioglioma.
Other glioneuronal lesions such as focal cortical dysplasia, dysembryoplastic neuroepithelial tumors, and gangliogliomas have exhibited aberrant CD34-immunoreactivity, speculated to relate to undifferentiated neural progeny.[
Molecular pathology
MAP kinase pathway activation might be implicated in gangliogliomas with reports of positive BRAF V600E mutations in up to 67% in specimens from certain age groups and locations, specifically the pediatric population.[
CONCLUSION
Gangliocytomas of the CNS of adults are very rare. As with our patient, they can arise from unusual locations such as the lateral ventricles and present at an older age. It is important to keep a broad differential whenever a suspicious lesion is encountered, especially when the lesion can be cured surgically, given its low-grade pathology.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Altman NR. MR and CT characteristics of gangliocytoma: A rare cause of epilepsy in children. AJNR Am J Neuroradiol. 1988. 9: 917-21
2. Beal MF, Kleinman GM, Ojemann RG, Hochberg FH. Gangliocytoma of third ventricle: Hyperphagia, somnolence, and dementia. Neurology. 1981. 31: 1224-8
3. Becker AJ, Klein H, Baden T, Aigner L, Normann S, Elger CE. Mutational and expression analysis of the reelin pathway components CDK5 and doublecortin in gangliogliomas. Acta Neuropathol. 2002. 104: 403-8
4. Becker AJ.editors. Ganglioglioma and Gangliocytoma. WHO Classification of Tumours of the Central Nervous System. 2007. p. 103-5
5. Blümcke I, Wiestler OD. Gangliogliomas: An intriguing tumor entity associated with focal epilepsies. J Neuropathol Exp Neurol. 2002. 61: 575-84
6. Capper D, Becker AJ, Giannini C, Figarella-Branger D, Huse JT, Rosemblum MK.editors. Gangliocytoma. WHO Classification of Tumours of the Central Nervous System, Revised. Lyon: International Agency for Research on Cancer; 2016. p. 136
7. Cavanagh JB. On certain small tumours encountered in the temporal lobe. Brain. 1958. 81: 389-405
8. Chung SN, Rosemond RL, Graham D. Prenatal diagnosis of a fetal intracranial tumor. J Ultrasound Med. 1998. 17: 521-3
9. Ebina K, Suzuki S, Takahashi T, Iwabuchi T, Takei Y. Gangliocytoma of the pineal body a case report and review of the literature. Acta Neurochir. 1985. 74: 134-40
10. Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, Vinet MC. Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron. 1999. 23: 247-56
11. Hirose T, Scheithauer BW, Lopes MB, Gerber HA, Altermatt HJ, van den Berg SR. Ganglioglioma. Cancer. 1997. 79: 989-1003
12. Horská A, Barker PB. Imaging of brain tumors: MR spectroscopy and metabolic imaging. Neuroimaging Clin N Am. 2010. 20: 293-310
13. Itoh Y, Yagishita S, Chiba Y. Cerebral gangliocytoma. Acta Neuropathol. 1987. 74: 169-78
14. Johannsson JH, Rekate HL, Roessmann U. Gangliogliomas: Pathological and clinical correlation. J Neurosurg. 1981. 54: 58-63
15. Kim HS, Lee HK, Jeong AK, Shin JH, Choi CG, Khang SK. Supratentorial gangliocytoma mimicking extra-axial tumor: A report of two cases. Korean J Radiol. 2001. 2: 108-12
16. Komotar RJ, O’Toole JE, Mocco J, Khandji AG, Keller CE, Connolly ES. Gangliocytoma of the spinal cord. Neurosurgery. 2007. 60: 895-900
17. Love S, Louis D, Ellison DW. Greenfield’s Neuropathology. Boca Raton, Florida, United States: CRC Press; 2008. 2:
18. Menon G, Patro SN, Krishnakumar K, Kesavadas C, Nair S, Radhakrishnan VV. Subfrontal gangliocytoma masquerading as olfactory groove meningioma. Br J Neurosurg. 2009. 23: 79-82
19. Mirzai H, Isisag A, Selcuki M. Incidental gangliocytoma in a middle-age adult: A case report. Ann Neurosurg. 2004. 4: 1-6
20. Mohila CA, Rauch RA, Adesina AM, Adesina AM, Tihan T, Fuller CE, Poussaint TY.editors. Gangliocytoma and ganglioglioma. Atlas of Pediatric Brain Tumors. Cham. Springer; 2016. p. 185-94
21. Nelson JS.editors. Ganglioglioma and gangliocytoma. Pathology and Genetics of Tumours of the Nervous System. Berlin, Germany: Springer; 2000. p. 96-8
22. Nikolic M, Chou MM, Lu W, Mayer BJ, Tsai LH. The p35/ Cdk5 kinase is a neuron-specific Rac effector that inhibits Pak1 activity. Nature. 1998. 395: 194-8
23. Pekmezci M, Villanueva-Meyer JE, Goode B, van Ziffle J, Onodera C, Grenert JP. The genetic landscape of ganglioglioma. Acta Neuropathol Commun. 2018. 6: 47
24. Peretti-Viton P, Perez-Castillo AM, Raybaud C, Grisoli F, Bernard F, Poncet M. Magnetic resonance imaging in gangliogliomas and gangliocytomas of the nervous system. J Neuroradiol. 1991. 18: 189-99
25. Prayson RA, Khajavi K, Comair YG. Cortical architectural abnormalities and MIB1 immunoreactivity in gangliogliomas. J Neuropathol Exp Neurol. 1995. 54: 513-20
26. Ratilal B, McEvoy A, Sisodiya S, Thom M, Toma A. Diffuse cerebral gangliocytoma in an adult with late-onset refractory epilepsy. Neuropathol Appl Neurobiol. 2007. 33: 706-9
27. Rubinstein LJ.editors. Tumors of the Central Nervous System. Washington, DC: AFIP; 1972. p. 349-60
28. Russo CP, Katz DS, Corona RJ, Winfield JA. Gangliocytoma of the cervicothoracic spinal cord. AJNR Am J Neuroradiol. 1995. 16: 889-91
29. Sherazi ZA. Gangliocytoma-magnetic resonance imaging characteristics. Singapore Med J. 1998. 39: 373-5
30. Shin JH, Lee HK, Khang SK, Kim DW, Jeong AK, Ahn KJ. Neuronal tumors of the central nervous system: Radiologic findings and pathologic correlation. Radiographics. 2002. 22: 1177-89
31. Thom M, Blümcke I, Aronica E. Long-term epilepsy-associated tumors: Long-term epilepsy associated tumors. Brain Pathol. 2012. 22: 350-79
32. Weis S, Sonnberger M, Dunzinger A, Voglmayr E, Aichholzer M, Kleiser R, Weis S, Sonnberger M, Dunzinger A, Voglmayr E, Aichholzer M, Kleiser R.editors. Ganglioglioma/gangliocytoma. Imaging Brain Diseases. A Neuroradiology Nuclear Medicine, Neurosurgery, Neuropathology and Molecular Biology-Based Approach. Vienna: Springer; 2019. p. 1553-65
33. Wen PY, Huse JT. 2016 World Health Organization classification of central nervous system tumors. Continuum (Minneap Minn). 2017. 23: 1531-47
34. Wolf HK, Müler MB, Spänle M, Zenther J, Schramm J, Wiestler OD. Ganglioglioma: A detailed histopathological and immunohistochemical analysis of 61 cases. Acta Neuropathol. 1994. 88: 166-73
35. Yagyu K, Sueda K, Shiraishi H, Asahina N, Sakurai K, Kohsaka S. Direct correlation between the facial nerve nucleus and hemifacial seizures associated with a gangliocytoma of the floor of the fourth ventricle: A case report. Epilepsia. 2011. 52: e204-6
36. Yamamoto T, Komori T, Shibata N, Toyoda C, Kobayashi M. Multifocal neurocytoma/gangliocytoma with extensive leptomeningeal dissemination in the brain and spinal cord. Am J Surg Pathol. 1996. 20: 363-70
37. Zulch KJ.editors. Histological typing of tumours of the central nervous system. International Histological Classification of Tumours No 21. 1979. p. 19-24

