

- Department of Internal Medicine, Division of Endocrinology, University of Cincinnati, Cincinnati, Ohio, Unite States.
- Department of Pathology and Laboratory Medicine, University of Cincinnati, Cincinnati, Ohio, Unite States.
- Department of Neurological Surgery, University of Louisville, Louisville, Kentucky, Unite States.
Correspondence Address:
Ruchi Bhabhra
Department of Internal Medicine, Division of Endocrinology, University of Cincinnati, Cincinnati, Ohio, Unite States.
DOI:10.25259/SNI_356_2019
Copyright: © 2019 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Yufei Dai, Matthew Hagen, Norberto Andaluz, Ruchi Bhabhra. Aggressive granular cell tumor of the neurohypophysis with optic tract edema and invasion into third ventricle. 15-Nov-2019;10:217
How to cite this URL: Yufei Dai, Matthew Hagen, Norberto Andaluz, Ruchi Bhabhra. Aggressive granular cell tumor of the neurohypophysis with optic tract edema and invasion into third ventricle. 15-Nov-2019;10:217. Available from: http://surgicalneurologyint.com/surgicalint-articles/9751/
Date of Submission
27-Sep-2019
Date of Acceptance
08-Oct-2019
Date of Web Publication
15-Nov-2019
Abstract
Background: Granular cell tumors (GCTs) of the neurohypophysis are parasellar tumors arising from pituicytes in the neurohypophysis and are generally considered benign slow-growing tumors. We present a case of sellar GCT with aggressive features.
Case Description: A 70-year-old female presented with progressive vision impairment found to have bitemporal visual field defects. Subsequent magnetic resonance imaging (MRI) revealed a 2.9 cm × 2.5 cm × 2.5 cm parasellar mass with extension into the third ventricle and causing optic tract edema (OTE). Right frontotemporal orbital craniotomy was performed and the tumor was partially removed to decompress optic nerves. Pathology identified the tumor as granular tumor of the sellar region. The patient’s vision improved minimally after the surgery. Follow-up MRI after 3 months and 11 months showed stable left OTE.
Conclusion: GCTs were thought to be benign tumors with slow growth, but they could potentially possess aggressive features and invade into surrounding structures as described in this case. OTE can be a rare MRI finding of GCTs. Only one case of GCT-related OTE has been reported in literature to our best knowledge.
Keywords: Granular cell tumor, Optic tract edema, Parasellar tumor, Third ventricle invasion
INTRODUCTION
Granular cell tumors (GCTs) of the neurohypophysis are benign tumors arising from pituicytes in the neurohypophysis. It was first described by Boyce and Beadles as an infundibular mass in 1893 and was identified as a tumor with granular cell histology by Sternberg 30 years later.[
CASE REPORT
A 70-year-old female with progressive vision impairment and headaches of at least 6 months duration presented to her ophthalmologist for eyeglasses replacement. She was found to have bitemporal visual field defects, left worse than right. She had no relevant medical history and physical exam was unremarkable except impaired vision. The patient had no significant symptoms of hypopituitarism and pituitary function was not evaluated by a neuroendocrinologist at this time.
Magnetic resonance imaging (MRI) of the brain with contrast revealed a 2.9 cm × 2.5 cm × 2.5 cm well-defined parasellar mass in the region of the hypothalamus and optic chiasm. The lesion was isointense on precontrast T1 and fluid-attenuated inversion recovery (FLAIR), hypointense on T2 [
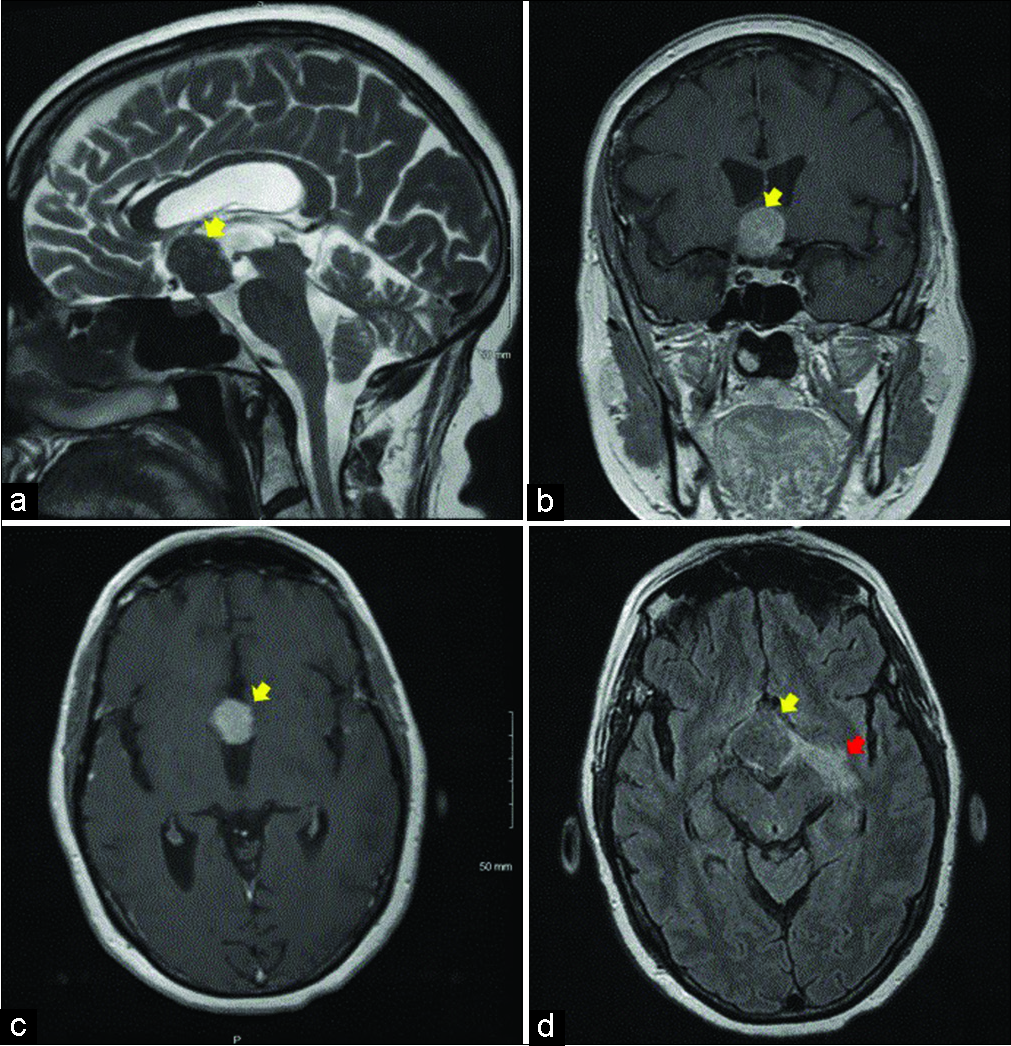
Based on these findings, the initial differential diagnosis included hypothalamic chiasmatic glioma, choristoma and less likely lymphoma. The patient underwent right frontotemporal orbital craniotomy with intraoperative neurophysiologic monitoring and image-guided neuronavigation for partial resection of the tumor and decompression of the bilateral optic nerves. Intraoperatively, a large suprasellar mass extending over the optic chiasm was identified. The optic chiasm was almost inseparable from, hard to define from the tumor. Frozen section histology was consistent with possible GCT or less likely plasmacytoma. Given the pathologic diagnosis, debulking of the tumor was completed in order to decompress the optic nerves.
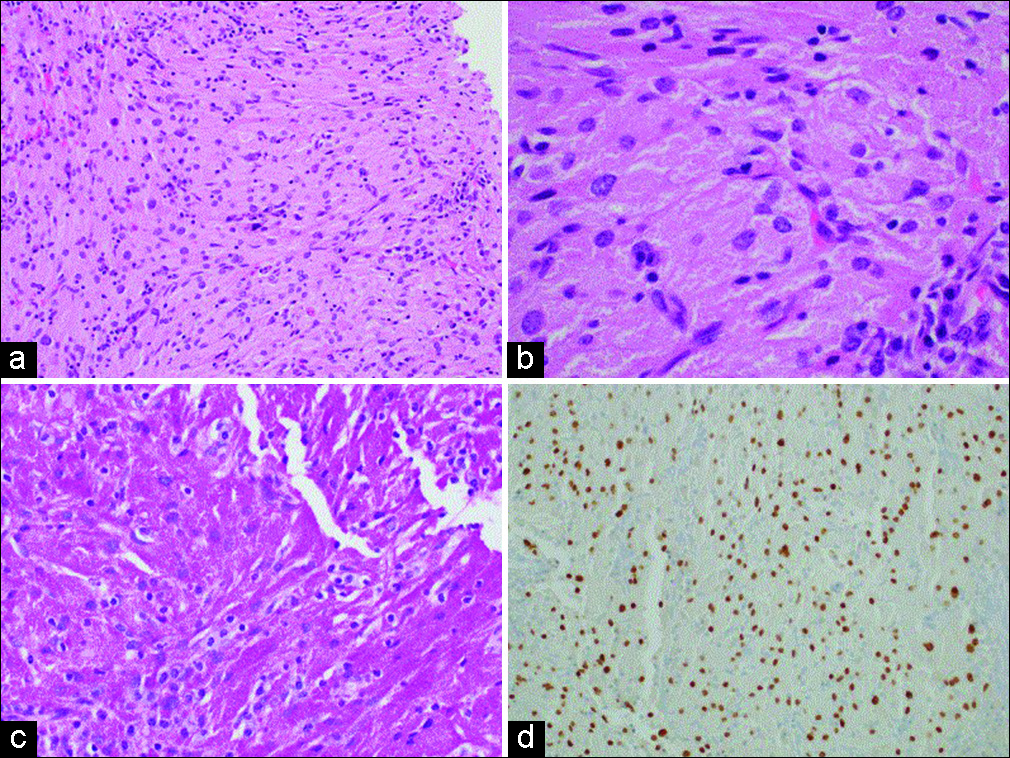
The formal pathology report identified the tumor as GCT of the sellar region, World Health Organization (WHO) Grade I. The tumor consisted of epithelioid to vaguely spindled cells with round to ovoid nuclei and abundant granular eosinophilic cytoplasm [
Immediate postoperative MRI showed a 2.6 cm × 2.4 cm × 2.4 cm anteriorly resected mass in close proximity to the optic chiasm. The patient was discharged home on postoperative day 3 with no new neurologic deficits. Her vision improved minimally after the surgery. MRI 3 months and 11 months after the surgery showed that the residual mass was stable in size. There was persistent abnormal T2-FLAIR signal hyperintensity in the left optic radiation consistent with persistent edema along the left optic tract. She did not develop diabetes insipidus after the surgery but she complained of excessive fatigue at 5 months follow-up. Evaluation of pituitary function at the time showed a low normal free thyroxine at 0.63 ng/dL (normal range: 0.61−1.76 ng/dL) and a low normal morning cortisol at 8.9 µg/dL (normal range: 6.7–22.6 µg/dL). Her insulin-like gorwth factor-1, thyroid-stimulating hormone, and adrenocorticotropic hormone (ACTH) was normal. She was placed on levothyroxine and hydrocortisone replacement due to the concern of central hypothyroidism and adrenal insufficiency. Hydrocortisone was discontinued after 3 weeks due to a normal ACTH stimulation test.
DISCUSSION
Symptomatic GCTs of the neurohypophysis are rare tumors of the sellar region. GCT of the neurohypophysis was classified as a benign tumor according to the 2016 WHO classification of the central nervous system tumors.[
Symptomatic GCTs are very rare with <100 cases reported in literature.[
Covington et al.[
In addition to typical MRI features of GCTs, increased signal along bilateral optic tracts and visualized portion of the optic chiasm consistent with OTE was observed in this patient. OTE was thought to be specific to craniopharyngiomas only, but it is currently considered to be caused by various parasellar and sellar lesions. The mechanism underlying the development of OTE is not totally clear though a few hypotheses were proposed. In patients with craniopharyngioma, leakage of cyst contents could potentially cause local inflammation and edema, whereas in patients with other types of sellar or parasellar lesions, local edema, and OTE were attributed to direct tumor invasion and retention of interstitial fluid in perivascular spaces caused by tumor blockage.[
The exact diagnosis of GCTs depends on pathological examination.[
GCTs are generally considered benign and slow-growing tumors with no pronounced tendency for invasion or recurrence.[
Surgical resection remains the mainstay of treatment to reduce the compression of surrounding structures. However, gross total resection is often prohibited by the firm and vascular nature of the GCTs along with lack of obvious dissection plane between and tumor and normal brain.[
CONCLUSION
We describe a patient with symptomatic GCT of neurohypophysis with OTE and third ventricle invasion, indicating that this benign tumor could potentially possess aggressive features.
Declaration of patient consent
Patient’s consent not obtained as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Baggenstos M, Chew E, Butman JA, Oldfield EH, Lonser RR. Progressive peritumoral edema defining the optic fibers and resulting in reversible visual loss. J Neurosurg. 2008. 109: 313-7
2. Barrande G, Kujas M, Gancel A, Turpin G, Bruckert E, Kuhn JM. Granular cell tumors. Rare tumors of the neurohypophysis. Presse Med. 1995. 24: 1376-80
3. Bussat A, Proisy M, Bruneau B, Bouzillé G, Chappé C, Riffaud L. Edema of the optic tract in patients with tumors of the sellar region: Clinical and visual implications in the pediatric population. J Neurosurg Pediatr. 2018. 21: 516-22
4. Cohen-Gadol AA, Pichelmann MA, Link MJ, Scheithauer BW, Krecke KN, Young WF. Granular cell tumor of the sellar and suprasellar region: Clinicopathologic study of 11 cases and literature review. Mayo Clin Proc. 2003. 78: 567-73
5. Covington MF, Chin SS, Osborn AG. Pituicytoma, spindle cell oncocytoma, and granular cell tumor: Clarification and meta-analysis of the world literature since 1893. AJNR Am J Neuroradiol. 2011. 32: 2067-72
6. Han F, Gao L, Wang Y, Jin Y, Lv Y, Yao Z. Clinical and imaging features of granular cell tumor of the neurohypophysis: A retrospective analysis. Medicine (Baltimore). 2018. 97: e9745-
7. Ju DG, Jeon C, Kim KH, Park KA, Hong SD, Seoul HJ.editors. Clinical significance of tumor-related edema of optic tract affecting visual function in patients with sellar and suprasellar tumors. World Neurosurg. 2019. p.
8. Liu HL, Huang BY, Zhang MS, Wang HR, Qu YM, Yu CJ. Sellar and suprasellar granular cell tumor of neurohypophysis. Chin Med J (Engl). 2017. 130: 741-3
9. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016. 131: 803-20
10. Saeki N, Nagai Y, Matsuura I, Uchino Y, Kubota M, Murai H. Histologic characteristics of normal perivascular spaces along the optic tract: New pathogenetic mechanism for edema in tumors in the pituitary region. AJNR Am J Neuroradiol. 2004. 25: 1218-22
11. Saeki N, Uchino Y, Murai H, Kubota M, Isobe K, Uno T. MR imaging study of edema-like change along the optic tract in patients with pituitary region tumors. AJNR Am J Neuroradiol. 2003. 24: 336-42
12. Schaller B, Kirsch E, Tolnay M, Mindermann T. Symptomatic granular cell tumor of the pituitary gland: Case report and review of the literature. Neurosurgery. 1998. 42: 166-70
13. Shizukuishi T, Abe O, Haradome H, Fukushima T, Katayama Y, Sugitani M. Granular cell tumor of the neurohypophysis with optic tract edema. Jpn J Radiol. 2014. 32: 179-82
14. Shuangshoti S, Chantra K, Navalitloha Y, Charoonwatanalaoha S, Shuangshoti S. Atypical granular cell tumor of the neurohypophysis: A case report with review of the literature. J Med Assoc Thai. 1998. 81: 641-6
15. Tomita T, Gates E. Pituitary adenomas and granular cell tumors. Incidence, cell type, and location of tumor in 100 pituitary glands at autopsy. Am J Clin Pathol. 1999. 111: 817-25

