

- Department of Neurosurgery, Bombay Hospital Institute of Medical Sciences, Mumbai, Maharashtra, India.
- Department of Radiology, Bombay Hospital Institute of Medical Sciences, Mumbai, Maharashtra, India.
Correspondence Address:
Chandan Mohanty, Department of Neurosurgery, Bombay Hospital Institute of Medical Sciences, Mumbai, Maharashtra, India.
DOI:10.25259/SNI_581_2022
Copyright: © 2022 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Chandan Mohanty1, Kalp Shandilya1, Chandrasekhar Eknath Deopujari1, Gaurav Gupta1, Vikram Karmarkar1, Sunila Jaggi2. Cervicomedullary glioblastoma: A report of two cases with review of literature. 16-Dec-2022;13:579
How to cite this URL: Chandan Mohanty1, Kalp Shandilya1, Chandrasekhar Eknath Deopujari1, Gaurav Gupta1, Vikram Karmarkar1, Sunila Jaggi2. Cervicomedullary glioblastoma: A report of two cases with review of literature. 16-Dec-2022;13:579. Available from: https://surgicalneurologyint.com/surgicalint-articles/12062/
Date of Submission
28-Jun-2022
Date of Acceptance
26-Nov-2022
Date of Web Publication
16-Dec-2022
Abstract
Background: Cervicomedullary glioblastoma is an extremely rare clinical entity and the principles of its management are not well understood.
Case Description: We report two cases of cervicomedullary glioblastoma in young patients aged 12 and 30 years with contrasting clinical presentation and outcomes. The 12-year-old child had rapid onset bulbar symptoms, with frank infiltration of the medulla due to which the patient succumbed within 4 weeks of surgery. The 30-year-old adult had a relatively slow disease onset and progression and made a good neurological recovery without disease progression at 16 months after surgery. To the best of our knowledge, we also report only the second adult patient in the literature with a dorsally exophytic cervicomedullary glioblastoma. Difficulties in diagnosis and management are discussed with a review of the pertinent literature.
Conclusion: The overall outcome depends on the rapid progression and severity of preoperative symptoms and the degree of tumor infiltration noted in imaging and during surgery.
Keywords: Cervicomedullary, Exophytic, Glioblastoma, Intramedullary, Spinal cord
INTRODUCTION
Intra-axial cervicomedullary tumors (CMTs) are a heterogeneous group of tumors. CMT is usually a low-grade tumor with only a small fraction being malignant.[
CASE DESCRIPTION
Case 1
A 30-year-old female presented with numbness and heaviness left upper limb associated with a weak grip left hand for 6 months. She also complained of pain in the neck on and off for 1 year. On examination, she had weakness of the left shoulder with a weak left-hand grip. Her left shoulder power was 4/5 while her left-hand grip was approximately 80% of her contralateral normal hand without any other neurological deficits. Magnetic resonance imaging (MRI) of the cervical spine revealed an elongated, bulky, and expansile mass lesion extending from the superior margin of the medulla with a dorsally exophytic component, and caudally was up to C6 vertebral level [
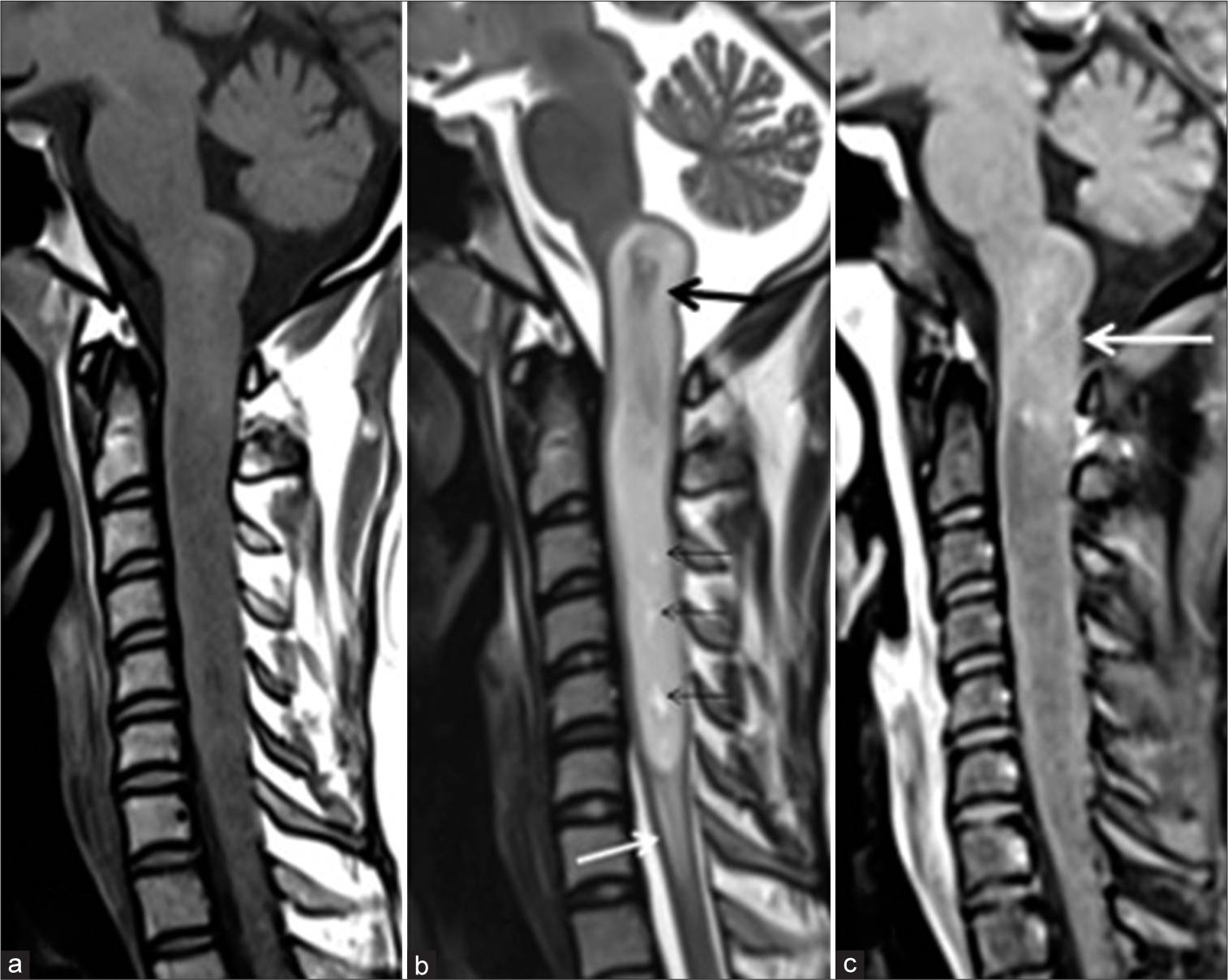
Figure 1:
Case 1. Sagittal T1-weighted (a), T2-weighted (b), and post contrast (c) images of the cervical spine show presence of a bulky, expansile, and elongated mass in the medulla and spinal cord upto the C6 vertebral level. Intrinsic T2 hypointensity in the cranial portion (black arrow), tiny cystic foci (thin black arrows), and hyperintense edema (white arrow) are seen in (b). Minimal patchy enhancement is seen in (c) (white arrow).
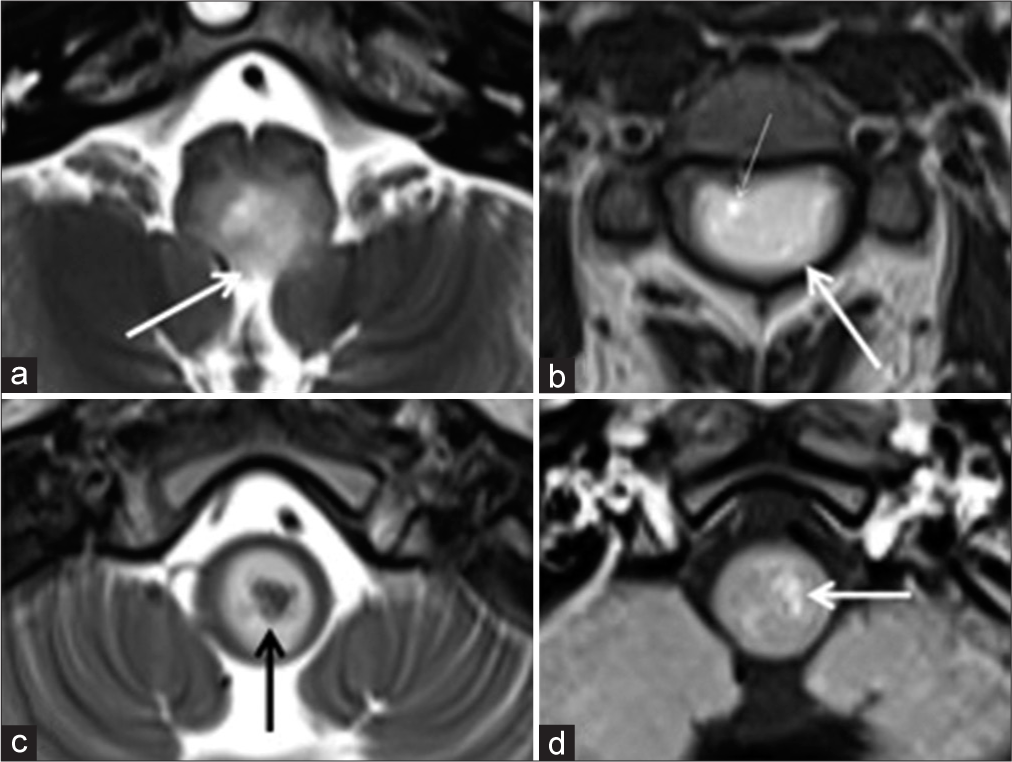
Figure 2:
Case 1. Axial T2-weighted (a-c) and post contrast T1-weighted (d) images reveal dorsal exophytic extension in (a) (white arrow), asymmetric involvement, more in the left hemicord (long white arrow), and cystic focus (thin white arrow) in (b), intrinsic T2 hypointensity in (c) (black arrow) and minimal enhancement in (d) (white arrow).
Case 2
A 12-year female presented with a complaint of acute onset weakness of the right upper and lower limb for 10 days associated with difficulty swallowing liquids and solids. It started with proximal weakness in the right upper limb and then progressed distally up to the hands affecting the grip and also the right lower limb. Power in the right upper limb proximally was 0/5, while the power in the right upper limb distally was 2/5. Her power in the right lower limb was 3/5 with grossly reduced sensations on the right side. Her bowel and bladder functions were intact; however, her gag reflex and cough reflex were weak.
MR imaging showed an expansile intramedullary lesion extending from the cervicomedullary junction to the C5 vertebral body appearing heterogeneously hyperintense on the T2W image and isointense on the T1W image [
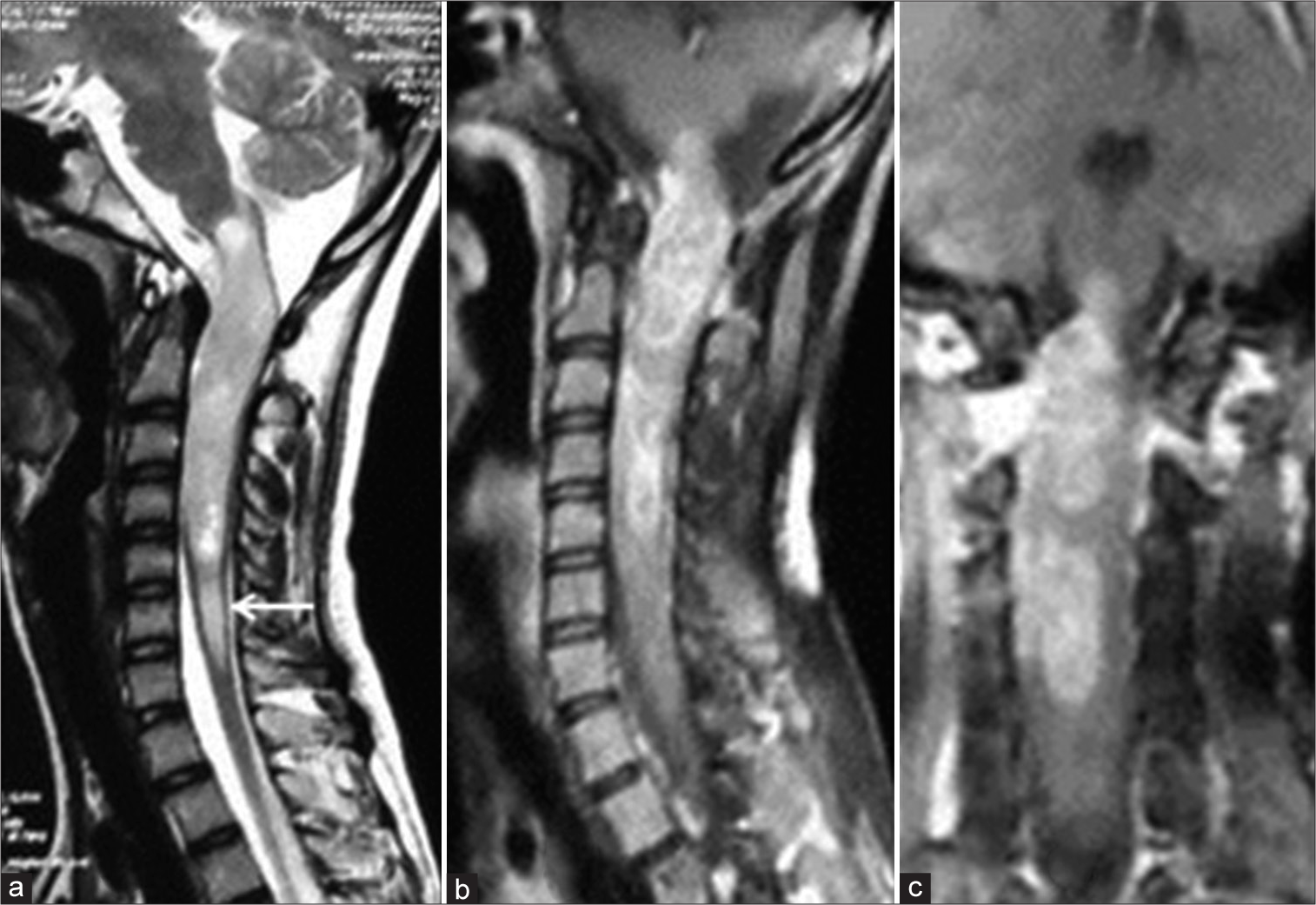
Based on imaging findings, suboccipital craniotomy and C1-C6 laminoplasty were done. Tumor infiltration was noted in the right cervical hemicord and medulla. About 90–95% of the tumor could be excised with residual left at the areas of clear infiltration which was confirmed with a postoperative MRI [
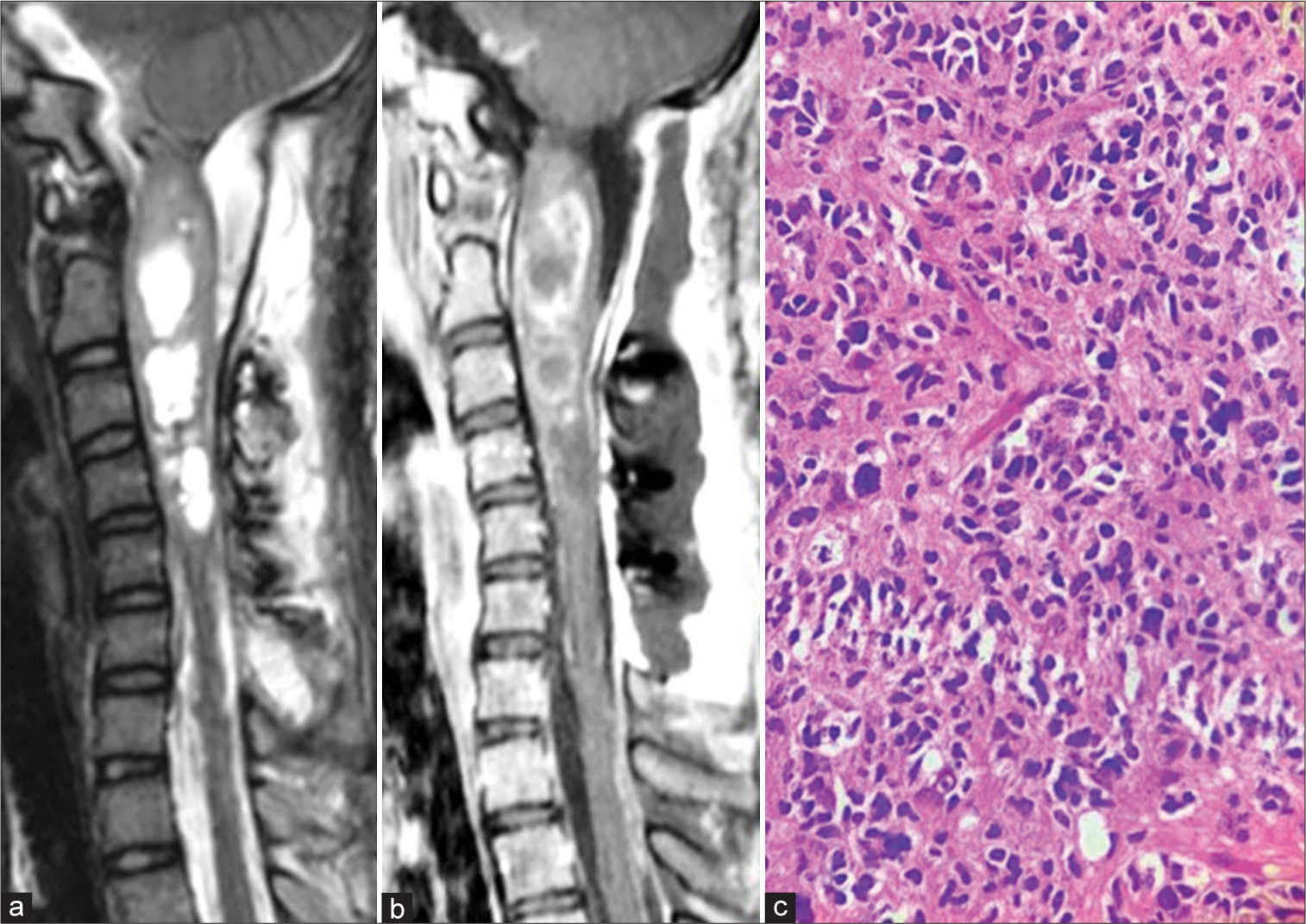
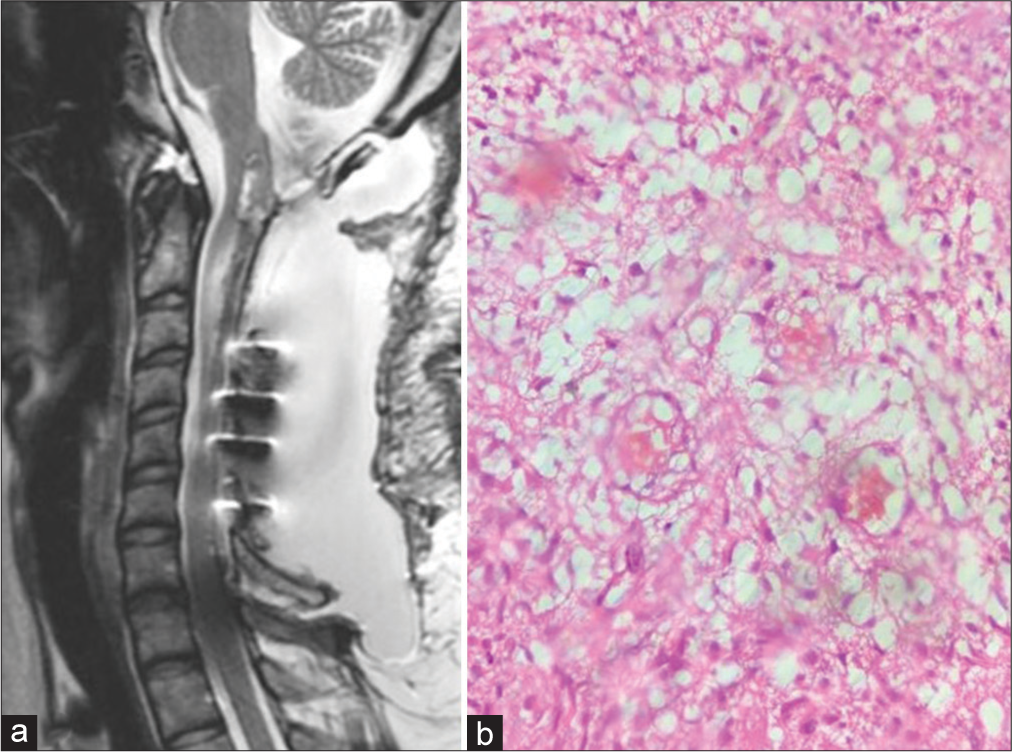
Figure 5:
Case 2. Sagittal T2-weighted (a) and post contrast T1-weighted (b) images of the cervical spine reveal marked excision of the mass lesion with postoperative changes. (c) Histopathology showing neurofibrillary background and pleomorphic cells suggestive of glioblastoma (Hematoxylin and Eosin, ×40 magnification).
DISCUSSION
In this paper, we describe two patients with cervicomedullary glioblastoma with contrasting clinical presentation and outcomes. The adult patient with a slower disease progression, without tumour infiltration into the medulla or cord was amenable for complete tumor resection and thus lead to a good neurological outcome. In contrast, the pediatric patient had a rapid onset of symptoms, with tumor infiltration into the medulla and cord, which allowed only a subtotal excision. This patient succumbed to her disease within 4 weeks of surgery.
Epstein classified brain stem tumors as diffuse, focal, and cervicomedullary.[
A study of 20 patients by Epstein and Wisoff suggested that cervicomedullary neoplasms were often “benign” and amenable to surgery.[
Samartzis et al. observed that astrocytomas have an association with NF-1 and are found mainly in males.[
Landi et al. in their analysis of intramedullary tumors of cervicomedullary regions observed cervicomedullary tumors are low-grade tumors affecting mainly the pediatric population.[
Histological type and grade are the most important determinants of prognosis in intramedullary spinal tumors.[
Radical surgical excision of CMT, in general, is the preferred treatment modality.[
The prognosis of these patients, however, is poor in spite of surgery and radiation therapy. This may be due to limitations in the current techniques to accurately determine the true extent of the dissemination of the disease. Based on the contrasting overall outcome of our two cases, it is reasonable to hypothesize that the rapidity, severity of the symptom onset, and the degree of tumor infiltration are important determinants of overall outcome in cervicomedullary glioblastoma.
CONCLUSION
Cervicomedullary glioblastoma in young patients is an extremely rare entity. The overall outcome depends on the rapid progression and severity of preoperative symptoms and the degree of tumor infiltration noted in imaging and during surgery.
Declaration of patient consent
Patients’ consent not required as patients’ identities were not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
1. Abbot R, Ragheb J, Epstein F, Cheek WR, Marlin AE, McLone DG, Reigel DH, Walker ML, editors. Brainstem tumors, Surgical indications. Pediatric Neurosurgery, Surgery of the Developing Nervous System. Philadelphia (PA): Saunders; 1994. p. 374-82
2. Ahmed R, Menezes AH, Awe OO, Mahaney KB, Torner JC, Weinstein SL. Long-term incidence and risk factors for development of spinal deformity following resection of pediatric intramedullary spinal cord tumors. J Neurosurg Pediatr. 2014. 13: 613-21
3. Cheng JS, Ivan ME, Stapleton CJ, Quinones-Hinojosa A, Gupta N, Auguste KI. Intraoperative changes in transcranial motor evoked potentials and somatosensory evoked potentials predicting outcome in children with intramedullary spinal cord tumors. J Neurosurg Pediatr. 2014. 13: 591-9
4. Daumas-Duport C, Appuzzo LJ, editors. Patterns of tumor growth and problems associated with histological typing of low-grade gliomas. Benign Cerebral Gliomas. Park Ridge: AANS; 1995. p. 125-47
5. Epstein FJ, Farmer J. Brain-stem glioma growth patterns. J Neurosurg. 1993. 78: 408-12
6. Epstein F, Wisoff J. Intra-axial tumors of the cervicomedullary junction. J Neurosurg. 1987. 67: 483-7
7. Fulton DS, Levin VA, Wara WM, Edwards MS, Wilson CB. Chemotherapy of pediatric brain-stem tumors. J Neurosurg. 1981. 54: 721-5
8. Konar SK, Bir SC, Maiti TK, Nanda A. A systematic review of overall survival in pediatric primary glioblastoma multiforme of the spinal cord. J Neurosurg Pediatr. 2017. 19: 239-48
9. Landi A, Brunetto GM, Gregori F, Delfini R, Tessitore E, Dehdashti AR, Schonauer C, Thome C, editors. Intramedullary tumors of the cervicomedullary junction. Surgery of the Cranio-Vertebral Junction. Champaign: Springer International Publishing; 2020. p. 367-93
10. Lober R, Sharma S, Bell B, Free A, Figueroa R, Sheils CW. Pediatric primary intramedullary spinal cord glioblastoma. Rare Tumors. 2010. 2: e48
11. Minehan KJ, Shaw EG, Scheithauer BW, Davis DL, Onofrio BM. Spinal cord astrocytoma: Pathological and treatment considerations. J Neurosurg. 1995. 83: 590-5
12. Merchant TE, Nguyen D, Thompson SJ, Reardon DA, Kun LE, Sanford RA. High-grade pediatric spinal cord tumors. Pediatr Neurosurg. 1999. 30: 1-5
13. Nair AP, Mehrotra A, Das KK, Srivastava AK, Sahu RN, Kumar R. Clinico-radiological profile and nuances in the management of cervicomedullary junction intramedullary tumors. Asian J Neurosurg. 2014. 9: 21-8
14. Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2014-2018. Neuro Oncol. 2021. 23: iii1-iii105
15. Samartzis D, Gillis CC, Shih P, O’Toole JE, Fessler RG. Intramedullary spinal cord tumors: Part I-epidemiology, pathophysiology, and diagnosis. Global Spine J. 2015. 5: 425-35
16. Smith RR, Zimmerman RA, Packer RJ, Hackney DB, Bilaniuk LT, Sutton LN. Pediatric brainstem glioma. Post-radiation clinical and MR follow-up. Neuroradiology. 1990. 32: 265-71
17. Tendulkar RD, Panandiker AS, Wu S, Kun LE, Broniscer A, Sanford RA. Irradiation of pediatric high-grade spinal cord tumors. Int J Radiat Oncol Biol Phys. 2010. 78: 1451-6
18. Warade AG, Misra BK. Dorsally exophytic cervicomedullary glioblastoma. J Clin Neurosci. 2014. 21: 1823-4
19. Weiner HL, Freed D, Woo HH, Rezai AR, Kim R, Epstein FJ. Intra-axial tumors of the cervicomedullary junction: Surgical results and long-term outcome. Pediatr Neurosurg. 1997. 27: 12-8

