

- Department of Neurosurgery, Aster MIMS Hospital, Kannur, Kerala, India.
Correspondence Address:
Shameej K. Vayalipath, Department of Neurosurgery, Aster MIMS Hospital, Kannur, Kerala, India.
DOI:10.25259/SNI_380_2022
Copyright: © 2022 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Ramesh C. Vasudevan, Shameej K. Vayalipath. Choroid plexus carcinoma in two siblings, with a novel genetic mutation in TP53 – A case report and review of literature. 26-Aug-2022;13:381
How to cite this URL: Ramesh C. Vasudevan, Shameej K. Vayalipath. Choroid plexus carcinoma in two siblings, with a novel genetic mutation in TP53 – A case report and review of literature. 26-Aug-2022;13:381. Available from: https://surgicalneurologyint.com/surgicalint-articles/11830/
Date of Submission
23-Apr-2022
Date of Acceptance
05-Aug-2022
Date of Web Publication
26-Aug-2022
Abstract
Background: Choroid plexus carcinoma (CPC) is an uncommon aggressive neuroectodermal-derived childhood brain malignancy with a dismal prognosis, especially when tumor protein p53 (TP53) mutations or malfunctions are present. The occurrence of these cancers is linked to germline and somatic anomalies at a number of genetic loci. We present a case report of CPC in two siblings which was found to be linked to a unique genetic mutation of TP53 in heterozygous state in both the father and the patient.
Case Description: A 2-year-old female child presented with a history of vomiting, headache, and seizures. A brain magnetic resonance imaging discovered a large-sized lesion in the left lateral ventricle with infiltration to surrounding brain parenchyma suggestive of aggressive choroid plexus neoplasm. Her only sibling (sister) died of CPC 1 year ago. Her parents are apparently healthy with no history of the central nervous system malignancies in the maternal and paternal sides. Since two children in a family were affected with CPC, genomic profiling of parents and patients was done. A novel frameshift variant c.72dupA,p. (Leu25Thrfs Ter4) was observed in exon 2 of TP53 in a heterozygous state in the proband. This variant was observed in her father in the heterozygous state.
Conclusion: CPC affecting siblings, associated with novel frameshift mutation in TP53 and inherited in an autosomal dominant pattern, is a rare entity. It has importance in genetic counseling and planning targeted molecular treatment. Genetic profiling is important for prognostication, as P53 pathway dysfunction carries a dismal prognosis, especially when it is associated with Li-Fraumeni syndrome.
Keywords: Choroid plexus carcinoma, Li-Fraumeni syndrome, Siblings, TP53 mutation
INTRODUCTION
Choroid plexus carcinomas (CPCs) are uncommon neuroectodermal tumors that makeup 10– 20% of intracranial cancers in children. They are aggressive tumors with a 0.3 case per million annual incidences.[
This case report is presented for three reasons. First, as per our knowledge is concerned, this is the first case report of CPC occurring in two siblings with the unique frameshift variation c.72dupA,p. (Leu25Thrfs Ter4) in TP53, which was inherited from the father in an autosomal dominant pattern. Second, it is important for implications in genetic counseling while planning the next pregnancy. Third, it is relevant in planning tailored individualized molecular treatment based on genetic abnormality.
CASE DESCRIPTION
A 2-year-old female child presented with a history of vomiting, headache, and seizures. A brain imaging (computed tomography and magnetic resonance imaging [MRI] scans) discovered a large-sized lesion in the left lateral ventricle with infiltration to surrounding brain parenchyma suggestive of aggressive choroid plexus neoplasm [
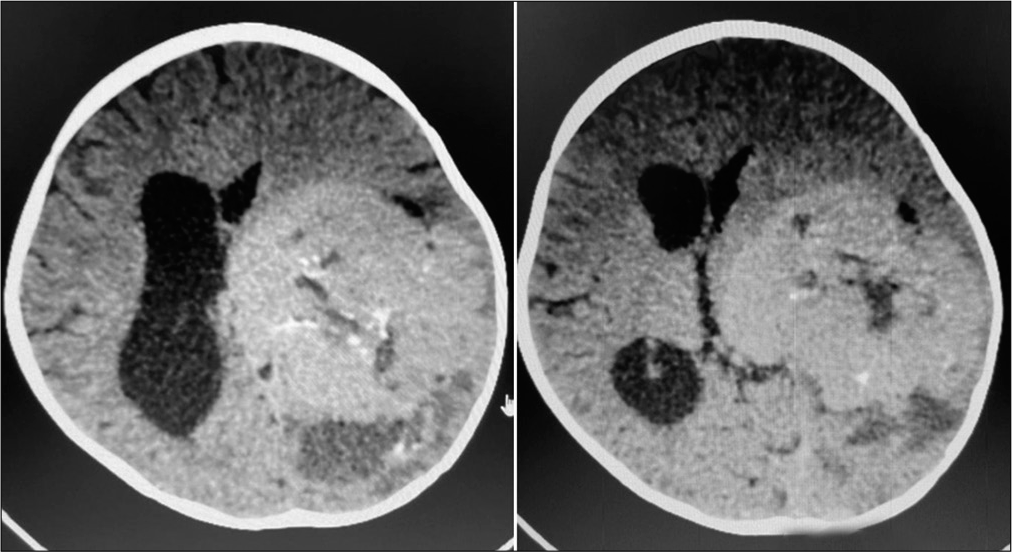
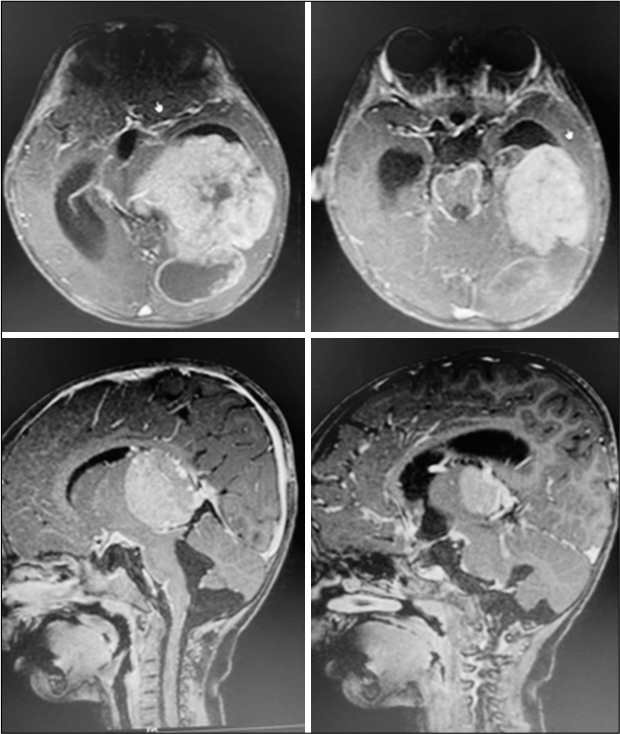
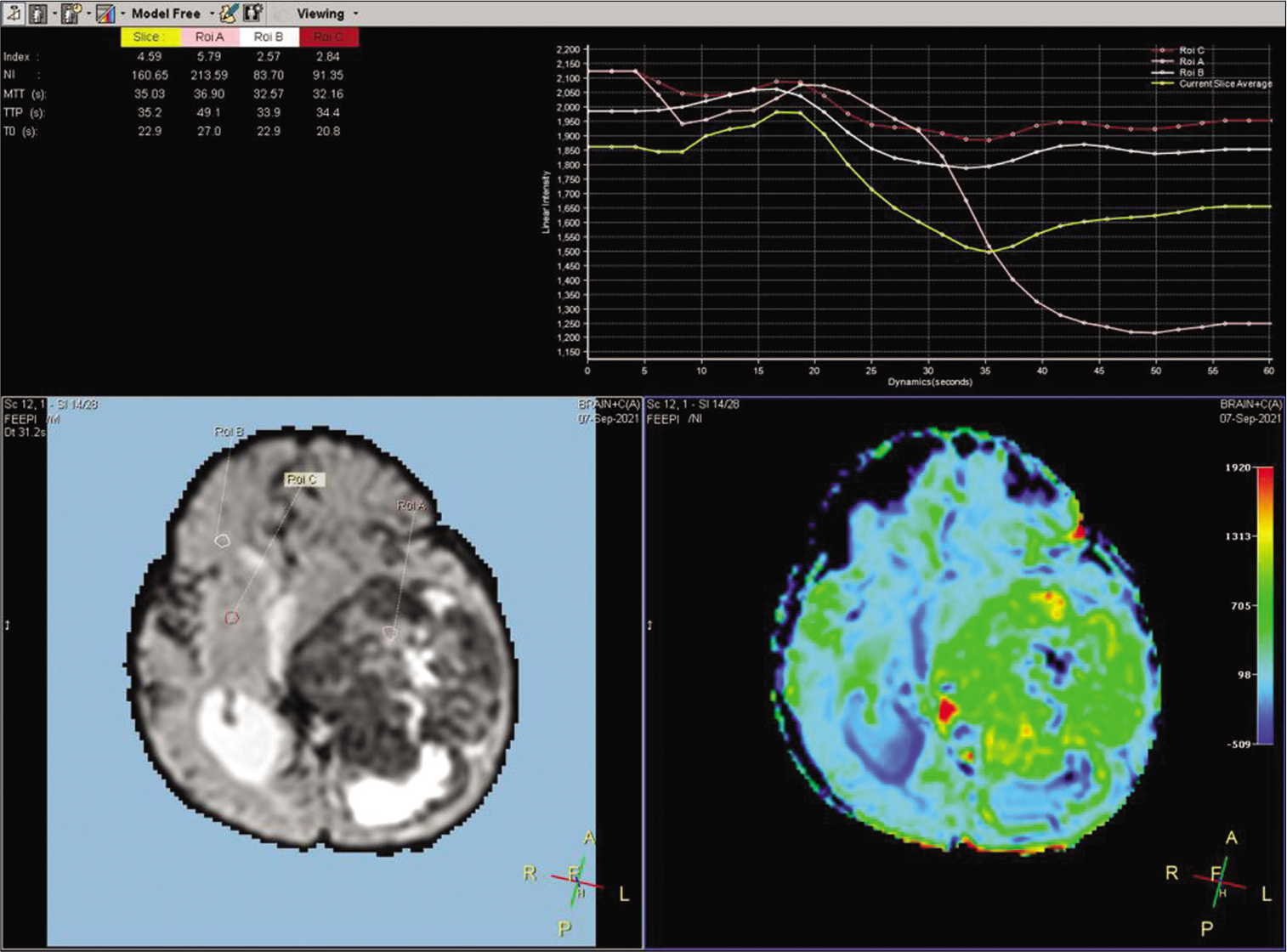
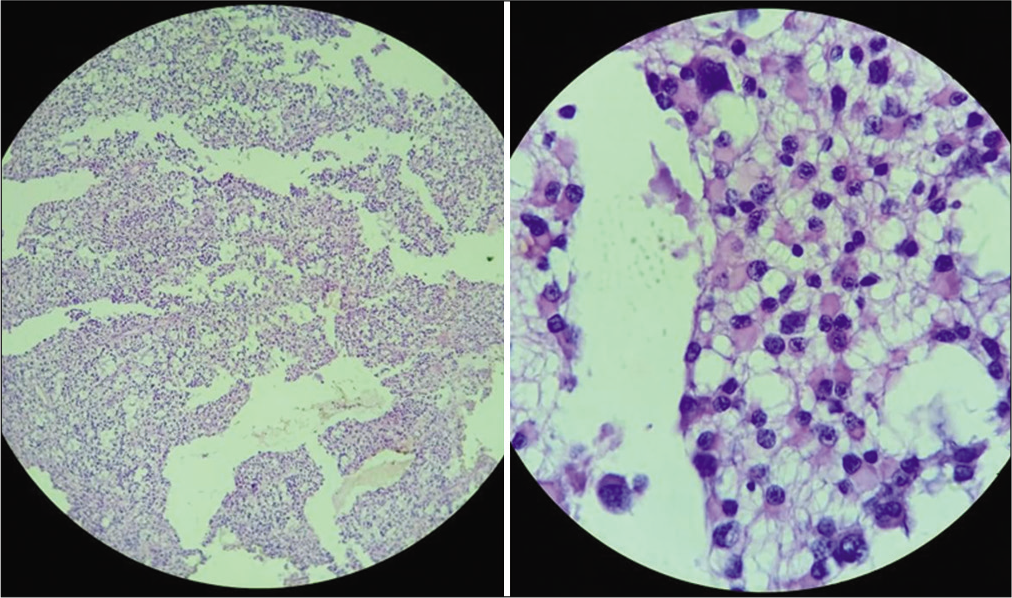
A novel frameshift variant c.72dupA,p. (Leu25Thrfs Ter4) was observed in exon 2 of TP53 in the heterozygous state in the proband; this variant was observed in her father in the heterozygous state. This was not observed in the mother and in the Genome Aggregation Database. It is noteworthy that the MRI or histopathological report of the elderly sibling was not available, and the parents had document about the CPC diagnosed and managed at some other hospital. Due to lack of availability of the tissue of elderly sibling, its genomic analysis could not be performed. Considering the Chompret criteria (2015), Li-Fraumeni syndrome is the likely cause for CPC in our cases.[
DISCUSSION
Although CPC is a rare pediatric brain neoplasm accounting for <1% of total cases of CNS tumors, CPC forms the classic brain tumor in patients with Li-Fraumeni syndrome.[
Genetic anomalies related to CPC
CPC s is among those CNS malignancies observed in LiFraumeni syndrome with TP53 germline mutations.[
Understanding the genetic mechanism of cancer by genomic sequencing and analysis could encourage the development of new medicines (therapies) or the repurposing of older medications.[
CONCLUSION
CPC is an aggressive and rare malignancy with a genetic predisposition. TP53 pathway deficiency/dysfunction is linked to a more aggressive nature and plays a crucial role in the formation of this illness. Here, we report a novel P53 frameshift mutation with an autosomal dominant pattern of inheritance from the father that resulted in CPC occurring in two daughters. Genetic workup of CPC is important in genetic and prenatal counseling and in prognostication. In such cases, it is important to be vigilant about the presence of Li-Fraumeni syndrome through the genetic workup so that individualized tailored molecular treatment regimens can be formulated.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
Dr. Amrut H Basava is acknowledged for language help and writing assistance.
References
1. Bettegowda C, Adogwa O, Mehta V, Chaichana KL, Weingart J, Carson BS. Treatment of choroid plexus tumors: A 20-year single institutional experience. J Neurosurg. 2012. 10: 398-405
2. Cornelius A, Foley J, Bond J, Nagulapally AB, Steinbrecher J, Hendricks WP. Molecular guided therapy provides sustained clinical response in refractory choroid plexus carcinoma. Front Pharmacol. 2017. 8: 652
3. Fang Z, Su Y, Sun H, Ge M, Qi Z, Hao C. Case report: Lifraumeni syndrome with central nervous system tumors in two siblings. BMC Pediatr. 2021. 21: 588
4. Gessi M, Giangaspero F, Pietsch T. Atypical teratoid/rhabdoid tumors and choroid plexus tumors: When genetics. “surprise” pathology. Brain Pathol. 2006. 13: 409-14
5. González MV, Pello MF, López-Larrea C, Suárez C, Menéndez MJ, Coto E. Loss of heterozygosity and mutation analysis of the p16 (9p21) and p53 (17p13) genes in squamous cell carcinoma of the head and neck. Clin Cancer Res. 1995. 1: 1043-9
6. Olivier M, Goldgar DE, Sodha N, Ohgaki H, Kleihues P, Hainaut P. Li-Fraumeni and related syndromes: Correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 2022. 63: 6643-50
7. Orr BA, Clay MR, Pinto EM, Kesserwan C. An update on the central nervous system manifestations of Li-Fraumeni syndrome. Acta Neuropathol. 2019. 139: 669-87
8. Rickert CH, Wiestler OD, Paulus W. Chromosomal imbalances in choroid plexus tumors. Am J Pathol. 2002. 160: 1105-13
9. Schneider K, Zelley K, Nichols KE, Garber J.editors. Li-Fraumeni syndrome. Gene Reviews®. Seattle, WA: University of Washington; 2019. p.
10. Sun MZ, Oh MC, Ivan ME, Kaur G, Safaee M, Kim JM. Current management of choroid plexus carcinomas. Neurosurg Rev. 2014. 37: 179-92
11. Tabori U, Shlien A, Baskin B, Levitt S, Ray P, Alon N. TP53 alterations determine clinical subgroups and survival of patients with choroid plexus tumors. J Clin Oncol. 2010. 28: 1995-2001
12. Tinat JBougeard GBaert-Desurmont SFrebourg T. Version of the Chompret Criteria for Li Fraumeni Syndrome. Available from: https://www.researchgate.net/publication/26714328_2009_version_of_the_chompret_criteria_for_li_fraumeni_syndrome [Last accessed on 2022 Jul 21].
13. Wang J, Merino DM, Light N, Murphy BL, Wang YD, Guo X. Myc and loss of p53 cooperate to drive formation of choroid plexus carcinoma. Cancer Res. 2019. 79: 2208-19
14. Wrede B, Liu P, Wolff JE. Chemotherapy improves the survival of patients with choroid plexus carcinoma: A meta-analysis of individual cases with choroid plexus tumors. J Neurooncol. 2007. 85: 345-51

