

- Department of Neurosurgery, International University of Health and Welfare Mita Hospital, Minato, Tokyo, Japan
- Department of Pathology, Tokai University School of Medicine, Isehara, Japan
- Division of Pathology, Nippon Koukan Hospital, Kanagawa, Japan
- Faculty of Health Sciences, Shonan University of Medical Science, Kanagawa, Japan
- Department of Neurosurgery, International University of Health and Welfare Narita Hospital, Narita, Japan.
Correspondence Address:
Ichiro Nakazato, Department of Neurosurgery, International University of Health and Welfare Mita Hospital, Tokyo, Japan.
DOI:10.25259/SNI_718_2023
Copyright: © 2023 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Ichiro Nakazato1, Kenichi Oyama1, Hisashi Ishikawa1, Yusuke Tabei1, Chie Inomoto2, Yoshiyuki Osamura3, Akira Teramoto4, Akira Matsuno5. Double pituitary neuroendocrine tumors in a patient with normal growth hormone level acromegaly: A case report and review of the literature. 15-Dec-2023;14:425
How to cite this URL: Ichiro Nakazato1, Kenichi Oyama1, Hisashi Ishikawa1, Yusuke Tabei1, Chie Inomoto2, Yoshiyuki Osamura3, Akira Teramoto4, Akira Matsuno5. Double pituitary neuroendocrine tumors in a patient with normal growth hormone level acromegaly: A case report and review of the literature. 15-Dec-2023;14:425. Available from: https://surgicalneurologyint.com/surgicalint-articles/12675/
Date of Submission
28-Aug-2023
Date of Acceptance
08-Nov-2023
Date of Web Publication
15-Dec-2023
Abstract
Background: Acromegaly is a rare disease caused by growth hormone (GH) hypersecretion caused by a pituitary neuroendocrine tumor (PitNET). However, some acromegaly patients show normal GH levels, and they can be a pitfall in clinical diagnosis. Moreover, rarely, synchronous true double or multiple PitNETs are encountered. Moreover, these PitNETs increase the risk of a left lesion during surgical exploration.
Case Description: The patient, who was a 73-year-old female, was referred to our hospital with a chief complaint of headache. Assessment of basal anterior pituitary function revealed a slightly high level of insulin-like growth factor-1 (IGF-1) (standard deviation, 2.4), and her physical findings exhibited mild acromegalic features. The endocrine evaluation confirmed acromegaly and magnetic resonance imaging (MRI) showed a macro PitNET with suprasellar extension. Endoscopic endonasal surgery (EES) was performed to remove the macro PitNET. Although postoperative MRI showed complete removal of the macro PitNET, endocrinological testing indicated no improvement in GH or IGF-1 excess. Pathological examination of the surgical specimen revealed a gonadotropic PitNET. Therefore, we repeated the MRI scan and found a micro PitNET in the thin left normal pituitary gland. A second EES was successfully performed to remove the micro PitNET completely, and both endocrinological and pathological examinations confirmed that the disease was cured.
Conclusion: Diagnosing acromegaly with low GH levels requires close monitoring. Double PitNETs are relatively rare and can cause incomplete remission of functional PitNETs.
Keywords: Acromegaly, Double pituitary neuroendocrine tumor, Endoscopic endonasal surgery, Pituitary, PitNET
INTRODUCTION
Pituitary neuroendocrine tumors (PitNETs) account for 10–15% of all intracranial tumors.[
Cases of acromegaly with normal or minimally elevated GH levels but abnormally high IGF-1 levels have been reported, and such “low GH acromegaly” can be a pitfall of clinical diagnosis.[
CASE DESCRIPTION
A 73-year-old female was referred to our hospital with a chief complaint of headache. She had a medical history of diabetes mellitus. Computed tomography and magnetic resonance imaging (MRI) scans revealed a macro PitNET (30 × 27 × 25 mm) [
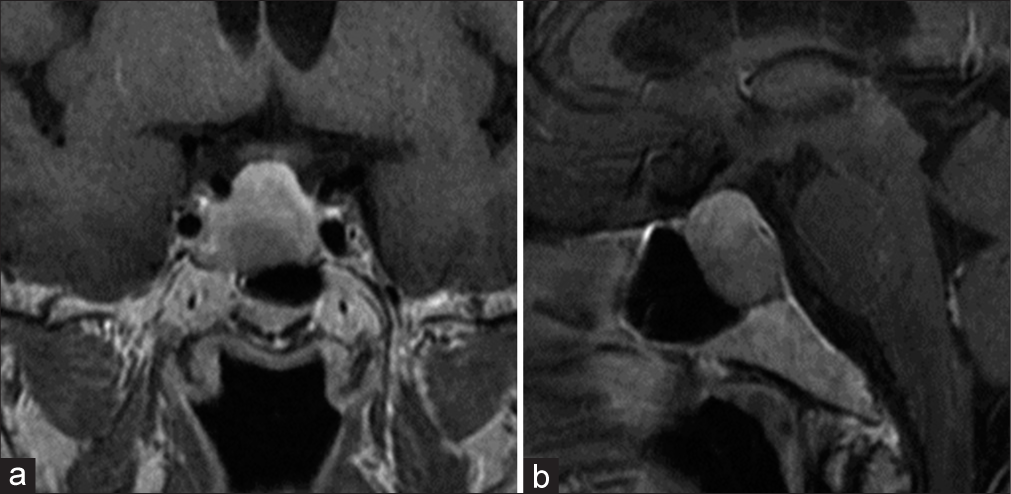
Laboratory findings indicated a normal level of GH: 2.02 ng/mL, normal values: 0.06–5.00 ng/mL, and other anterior pituitary hormones, except a slightly high level of IGF-1: 180 ng/mL, normal values: 54.00–167 ng/mL. Physical findings revealed characteristics of mild acromegaly, including mild facial and acral enlargement. Radiographic findings showed a cauliflower-like enlargement of the distal phalanx of the fingers and a heel pad thickness of 22.28 cm (normal values: <22 cm). Oral glucose tolerance test indicated insufficient suppression of GH (nadir GH: 1.29 ng/mL, normal values: <0.04 ng/mL), and the thyrotropin-releasing hormone stimulation test exhibited a paradoxical rise in serum GH level. These findings confirmed the diagnosis of acromegaly with low GH levels. Endoscopic endonasal surgery (EES) was performed to remove the macro PitNET. Although postoperative MRI showed complete tumor removal [
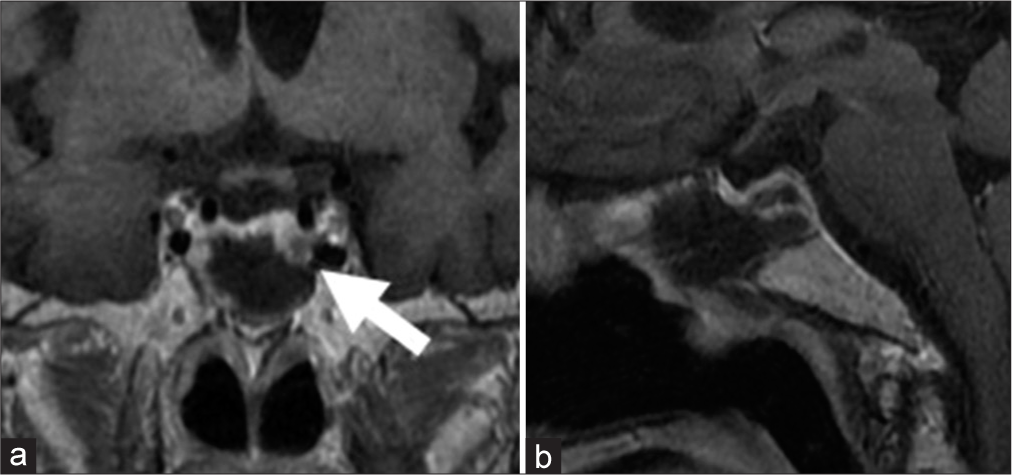
Figure 2:
(a) Magnetic resonance imaging (MRI) coronal section postoperative MRI reveals a complete removal of the macro pituitary neuroendocrine tumor (PitNET) and the residual micro PitNET (arrow) in the left thin, normal pituitary gland. (b) MRI sagittal section postoperative MRI reveals a complete removal of the macro PitNET.

Figure 3:
(a) Steroidogenic factor-1 (SF-1)-positive image immunohistochemical staining of the first surgical specimen shows positive staining for SF-1, follicle-stimulating hormone (FSH), and α-subunit, indicating that the resected tumor is a gonadotroph pituitary neuroendocrine tumor (PitNET), (b) FSH-positive image immunohistochemical staining of the first surgical specimen shows positive staining for SF-1, FSH, and α-subunit, indicating that the resected tumor is a gonadotroph PitNET, (c) α-subunit-positive image immunohistochemical staining of the first surgical specimen shows positive staining for SF-1, FSH, and α-subunit, indicating that the resected tumor is a gonadotroph PitNET.
The micro PitNET was successfully removed with a second EES [

Figure 4:
(a) Magnetic resonance imaging (MRI) coronal section MRI after the second operation shows the complete removal of the residual micro pituitary neuroendocrine tumor (PitNET) in the left thin, normal pituitary gland, (b) MRI sagittal section MRI after second operation shows the complete removal of the residual micro PitNET in the left thin, normal pituitary gland.

Figure 5:
(a) Growth hormone (GH)-Positive Image Immunohistochemical staining of the 2nd surgical specimen exhibits positive staining for GH, somatostatin receptor type 2 (SSTR-2), POU1F1/GHF-1(Pit-1), and α-subunit, which mean that the resected tumor is a somatotroph pituitary neuroendocrine tumor (PitNET), (b) SSTR2-positive image immunohistochemical staining of the 2nd surgical specimen exhibits positive staining for GH, SSTR-2, Pit-1, and α-subunit, which mean that the resected tumor is a somatotroph PitNET, (c) Pit-1-positive Image Immunohistochemical staining of the 2nd surgical specimen exhibits positive staining for GH, SSTR-2, Pit-1, and α-subunit, which mean that the resected tumor is a somatotroph PitNET, (d) α-subunit-Positive Image Immunohistochemical staining of the 2nd surgical specimen exhibits positive staining for GH, SSTR-2, Pit-1, and α-subunit, which mean that the resected tumor is a somatotroph PitNET.

DISCUSSION
The overall prevalence of “normal GH acromegaly,” whose serum GH level is within normal limits, is reported to be about 25–30% in all acromegaly.[
Some research has shown that the tumor size of normal-GH acromegaly is smaller than that of high-GH acromegaly.[
Sometimes, patients have double or multiple pituitary neuroendocrine tumors in the pituitary fossa.[
In this case, the patient had double PitNETs; one was a macro gonadotroph PitNET located in the center of the sella turcica, and the other was a micro somatotroph PitNET located in the thin, normal pituitary gland; therefore, we could not find the micro PitNET in the preoperative MRI before the 1st operation and failed to remove the responsible GH-producing tumor. The diagnosis of acromegaly was made based on elevated IGF-1 and clinical symptoms. Both histological findings and endocrinological examination allowed us to determine the failure of the 1st surgery, and we could then identify the residual responsible tumor using MRI.
CONCLUSION
Here, we report a case of double pituitary neuroendocrine tumors (PitNETs) in a patient with normal GH-level acromegaly. Double PitNETs may be a clinical pitfall, particularly in patients with endocrinopathy. If postoperative pituitary hormone excess does not improve after the complete removal of the tumor, we need to recheck pre and postoperative radiographic and pathological findings to explore the cause of surgical failure.
Ethical approval
Not applicable.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
1. Butz LB, Sullivan SE, Chandler WF, Barkan AL. “Micromegaly”: An update on the prevalence of acromegaly with apparently normal GH secretion in the modern era. Pituitary. 2016. 19: 547-51
2. Espinosa de Los Monteros AL, Sosa-Eroza E, Gonzalez B, Mendoza V, Mercado M. Prevalence, clinical and biochemical spectrum, and treatment outcome of acromegaly with normal basal GH at diagnosis. J Clin Endocrinol Metab. 2018. 103: 3919-24
3. Hashmi FA, Shamim MS. Pituitary adenoma: A review of existing classification systems based on anatomic extension and invasion. J Pak Med Assoc. 2020. 70: 368-70
4. Iacovazzo D, Bianchi A, Lugli F, Milardi D, Giampietro A, Lucci-Cordisco E. Double pituitary adenomas. Endocrine. 2013. 43: 452-7
5. Krug S, Boch M, Rexin P, Pfestroff A, Gress T, Michl P. Acromegaly in a patient with a pulmonary neuroendocrine tumor: Case report and review of current literature. BMC Res Notes. 2016. 9: 326
6. Magri F, Villa C, Locatelli D, Scagnelli P, Lagonigro MS, Morbini P. Prevalence of double pituitary adenomas in a surgical series: Clinical, histological and genetic features. J Endocrinol Invest. 2010. 33: 325-31
7. Patronas N, Bulakbasi N, Stratakis CA, Lafferty A, Oldfield EH, Doppman J. Spoiled gradient recalled acquisition in the steady state technique is superior to conventional postcontrast spin echo technique for magnetic resonance imaging detection of adrenocorticotropin-secreting pituitary tumors. J Clin Endocrinol Metab. 2003. 88: 1565-9
8. Pecorari IL, Mahali LP, Funari A, Fecher R, Suda N, Agarwal V. Silent corticotroph and somatotroph double pituitary adenoma: A case report and review of literature. J Neurol Surg Rep. 2022. 83: e33-8
9. Zieliński G, Maksymowicz M, Podgórski J, Olszewski WT. Double, synchronous pituitary adenomas causing acromegaly and Cushing’s disease. A case report and review of literature. Endocr Pathol. 2013. 24: 92-9
10. Zieliński G, Sajjad EA, Maksymowicz M, Pękul M, Koziarski A. Double pituitary adenomas in a large surgical series. Pituitary. 2019. 22: 620-32

