

- Department of Surgery, Section of Neurosurgery, Aga Khan University, Karachi, Pakistan
- Department of Pathology, Section of Histopathology, Aga Khan University, Karachi, Pakistan
Correspondence Address:
Fauzan Alam Hashmi
Department of Pathology, Section of Histopathology, Aga Khan University, Karachi, Pakistan
DOI:10.4103/2152-7806.169545
Copyright: © 2015 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Hashmi FA, Khan MF, Khan SA, Waqas M, Bari ME, Ahmed A. Ependymal tumors with oligodendroglioma like clear cells: Experience from a tertiary care hospital in Pakistan. Surg Neurol Int 16-Nov-2015;6:
How to cite this URL: Hashmi FA, Khan MF, Khan SA, Waqas M, Bari ME, Ahmed A. Ependymal tumors with oligodendroglioma like clear cells: Experience from a tertiary care hospital in Pakistan. Surg Neurol Int 16-Nov-2015;6:. Available from: http://surgicalneurologyint.com/surgicalint_articles/ependymal-tumors-with-oligodendroglioma-like-clear-cells/
Abstract
Background:Ependymal tumors with oligodendroglioma like clear cells have never been reported from Pakistan. We aimed to see the features and outcomes of this rare entity.
Methods:It was retrospective cohort conducted at the Department of Neurosurgery, Aga Khan University from 2003 to 2013. The medical records and radiology of patients with proven histopathology were reviewed. Analysis was done on SPSS 20.
Results:Eleven cases of ependymal tumors with clear cells were found, which equated to 1.5% of the total tumor burden in 11 years. The median age was 49 years. Most common presenting symptom was headache 54.5%. Out of 11 patients, 9 patients had a supratentorial tumor. Magnetic resonance imaging showed hypointense signals on T1 and hyperintense signals on T2-weighted images in all cases. Contrast enhancement was found in 9 patients (77.8%), necrosis and hemorrhage was found in 4 (36%) and 3 (27%) patients, respectively. Immunohistochemistry showed glial fibrillary acidic protein and epithelial membrane antigen positivity in all cases. Ki-67 showed high proliferative index in 6 patients. According to the World Health Organization grading of ependymal tumors, 2 patients had Grade II tumors, and 9 patients had Grade III tumors with clear cells. Gross total resection was achieved in 6 (54.5%) and subtotal resection in 5 patients (45.4%). Recurrence was observed in 9 patients. Six patients died of the disease. Median progression-free survival and overall survival was 8 months and 10 months, respectively.
Conclusion:Ependymal tumors with clear cells presented more commonly in Grade III lesions and were more aggressive in behavior with poorer outcome compared to similar studies.
Keywords: Anaplasia, clear cells, overall survival, progression-free survival, supratentorial
INTRODUCTION
Ependymal tumors are rare neuroepithilial tumor which represent only 2–8% of all central nervous system (CNS) malignancies.[
The literature has been scarce on this entity since it was first described in 1983.[
Grade II ependymoma undergo malignant transformation to Grade III and have lower rate of cure even with extensive resection, this is why although CCE is a variant of ependymoma Grade II, but most of the series published to date, show presence of clear cells more in histologically proven anaplastic ependymoma Grade III than Grade II.[
To the best of author's knowledge, there is no report on the behavior and outcome of disease from this country and hence we aimed to describe the frequency, clinical, radiological, surgical and histopathological aspects, and outcome of all ependymal tumors containing ODG like clear cells including WHO Grade II and Grade III tumors, managed at our institution along with a review of available literature.
MATERIALS AND METHODS
This is a retrospective case series of all ependymal tumors containing ODG like clear cells, surgically treated at the section of neurosurgery, Department of Surgery, Aga Khan University, Karachi, Pakistan from January 2003 to December 2013. Medical records of all primary brain neoplasms treated during this period were retrieved from the electronic database of the hospital using ICD code 190.0–191.9.
Data were systematically collected on a structured proforma, which included patient demographics, presenting complaints, duration of symptoms in months, Karnofsky performance score (KPS), radiology, course of treatment including surgery, adjuvant therapy (chemotherapy [CMT] and radiotherapy [RXT]), progression-free survival (PFS), and recurrence as suggested by interval 3 monthly magnetic resonance imaging (MRI) of brain and overall survival (OS).
Information about aforementioned aspects were obtained from patient initial out patient Department (OPD) notes, initial assessment forms, progress notes, operative notes retrieved through medical record office, and telephonic calls to patients/attendants were applicable. MRI and computerized tomography (CT) scans of brain were reviewed on picture archiving and communication system of the hospital (PACS) for the location of the tumor to lobe, initial size, and feature specific to tumors such as contrast enhancement, appearance on T1-weighted and T2-weighted images, and associated characteristics such as presence of mass effect, cystic, hemorrhagic, or necrotic areas were obtained. The histopathology slides were retrieved and reviewed and all the cases that contained the features of ependymal tumors with ODG like clear cells were described by a consultant histopathologist for features including the WHO grade and immunohistochemical stains for the purpose of current study.
Gross total resection (GTR) was defined as excision of more than 95% of tumor and subtotal resection (STR) was defined as excision of <95% tumor.
The data were recorded and analyzed using IBM-SPSS statistics 20 (SPSS 20, IBM, Armonk, NY, United States of America). Median was calculated for continuous variables and frequencies and proportions were calculated for categorical variables. Kaplan–Meier curve analysis was done for PFS and OS.
RESULTS
In an 11 year period from January 1, 2003, to December 31, 2013, there were a total of 727 cases of primary CNS tumors that were treated surgically at the Aga Khan University, of which there were 51 (7%) cases of ependymal tumors. Histopathologically proven clear cells were found in 11 (1.5%) cases.
The results showing age, gender, presenting symptoms, and duration of symptoms are summarized in

Imaging features
A total of 10 patients had MRI done, 1 patient had CT scans done for diagnosis and progression of the disease secondary to a cardiac pacemaker. The MRI features of all the patients in the series are summarized in
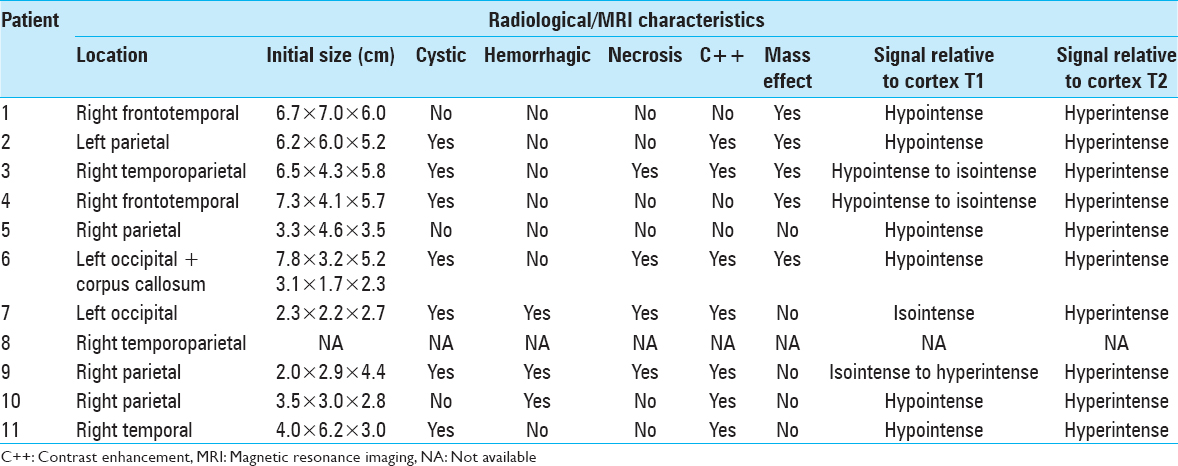
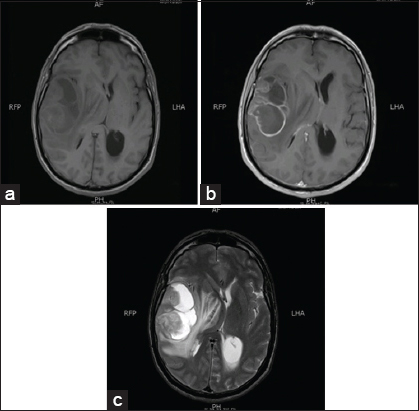
Figure 1
Magnetic resonance imaging scans of patient number 3.(a) T1-weighted magnetic resonance image of patient 3, showing hypo to isointense tumor in right temporoparietal cortex with compression and near effacement of right lateral ventricle, subfalcine herniation, and midline shift. (b) T1 with gadolinium of the same patient showing subtle peripheral ring enhancement and loculations.(c) T2-weighted image of the same patient showing cystic areas with solid component displaying hyperintense signals
Histopathology
The histopathological assessment showed diffuse sheets of spindle to epithelioid neoplastic cells embedded within a fibrillary background in all cases. The perivascular pseudorosettes were frequent in 8 cases and focal in 3. Clear ODG like cells, suggested by having moderate amount of clear to eosinophilic cytoplasm, moderate pleomorphic, round to oval nuclei surrounded by perinuclear halo were found in few areas in 5 cases, whereas more frequently in 6 cases. Increased cellularity, occasionally prominent nucleoli, atypical mitotic figures, necrosis, and microvascular proliferation was seen in 9 out of 11 patients. Minigemistocytes were seen in 2 cases.
Immunohistochemistry showed that the tumor was GFAP and EMA positive in all cases (100%). Ki-67 showed low proliferative index in 5 patients and high proliferative index in 6 patients. WHO class was Grade II in 2 patients and Grade III in 9 patients [
Figure 2
Histopathological slides of the patients in the series. (a) H and E stain of patient 1, showing clear cell morphology. (b) H and E stain of patient 1, showing perivascular pseudorosettes. (c) Slide displaying epithelial membrane antigen positivity in patient number 2.(d) Slide showing 10–15% (high) Ki-67 positivity in patient number 5
Treatment
Each case was discussed in the multidisciplinary tumor board meeting comprising of consultant neurosurgeons, radiologists, histopathologists, radio-oncologists, senior nurses, and medical oncologists. At the time of writing this article we are following the National Comprehensive Cancer Network (NCCN) USA, clinical practice guidelines in oncology for CNS cancers for the treatment of adult intracranial ependymoma.[
All 11 patients primarily underwent surgery, out of these 7 patients received concomitant external beam fractionated radiotherapy (EBRT) only, 3 patients received concomitant EBRT and CMT/temozolomide (TMZ), and 1 patient received no therapy (patient's wish) after initial diagnosis.
Recurrence was observed in 9 patients, 1 patient had no evidence of disease (NED) until writing this article, and 1 patient refused for further investigation or treatment. Out of 9 patients who had recurrence of disease, 6 patients did not have further treatment. In the remaining 3 patients repeat surgery was performed, of which 2 patients further received re-RXT and CMT (1 patient was given cisplatin plus etoposide 6 cycles, the other patient was given TMZ 6 cycles), whereas 1 patient received re-RXT alone.
Six patients expired of the disease. One patient refused for any further treatment post-GTR and expired 5 months after the primary surgery. Four underwent EBRT postprimary surgery, one of them remained progression free for almost 28 months postprimary surgery, underwent repeat surgery twice within an interval of 5 months followed by TMZ and re-RXT, and she expired on 52 months postprimary surgery. Three more underwent EBRT but had progression of disease, 2 patients underwent concomitant CMT (one with TMZ 6 cycles and other with cisplatin and etoposide 6 cycles) along with re-RXT. The patient with TMZ + re-RXT was dead of the disease 10 months postprimary surgery and patient with cisplatin plus etoposide + re-RXT had metastasis to corpus callosum, thalamus and had dural deposits, he was lost to follow-up at 19 months from primary surgery.
Currently, 4 patients are under follow-up until writing this article. One patient underwent STR, followed by concomitant TMZ, and EBRT had reduction in residual size and has no evidence of progression 10 months from the surgery. The second patient underwent GTR followed by EBRT in Italy, he remained progression free for 12 months however, the tumor recurred, he refused further treatment, he is alive on 67 weeks since his primary surgery; however, currently he is bedridden, dependent for all his activities, KPS 20. The third patient STR, her initial histology shows features of ODG, she was followed by TMZ and EBRT; however, her tumor recurred at 8 month postsurgery, she again underwent STR, this time the histopathology showed CCE, she underwent re-RXT, currently she has NED for 10 months post re-surgery, total 18 months from first surgery. The fourth patient underwent GTR + EBRT and had recurrence at 3 months and currently has stable disease at 5 months.
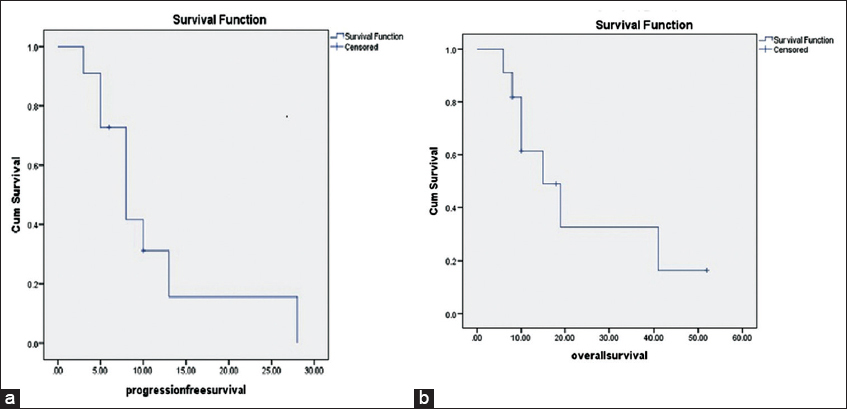
The median PFS was 8 months ranging from minimum 3 months and maximum 28 months, and the median OS was 10 months (range 6 months–52 months) after primary surgery. The extent of resection, intraoperative tumor findings, initial diagnosis, treatment given, recurrence, PFS, and OS are summarized in
DISCUSSION
CCE is considered to be Grade II supratentorial and extraventricular tumor, occurring predominantly in young patients.[
Interestingly, the median age of our series was 49 years (age range 24–72 years). This was significantly different from the cohorts of Fouladi et al.[
The treatment of intracranial ependymoma as suggested by NCCN guidelines 2014[
The clinical presentation is similar to that reported in other studies.[
With respect to radiological features, Fouladi et al.[
In our study, we observed hypointense signals on T1 and hyperintense signals on T2-weighted images, associated cysts, and enhancement in 9 out of 11 patients, mass effect was present in 5 patients; however, necrosis and hemorrhage were present in 4 and 3 patients, respectively.
Similar findings were reported by other authors.[
The gross intraoperative examination of the tumor is rarely reported in the literature, Jain et al.[
WHO has not clearly defined the histological criteria to classify the tumor as CCE with respect to the percentage of ODG like clear cells and ependymoma like areas present. Fouladi et al.[
The median PFS of our study was 8 ± 6.7 months postsurgery, and median OS was 10 ± 14 months postsurgery. This was significantly different from the cohorts of Kawano et al.,[
Our cohort highlights the significant amount of misdiagnosis and confusion in diagnosing CCE; this highlights the use of more precise immunohistochemical stains and cytogenetic testing to confirm the diagnosis. Preusser et al. described OLIG-2 an additional immunohistochemical marker to differentiate between ODG and CCE.[
Our limitation was a small sample size, a single institution data, and a retrospective study design. This is the first report from Pakistan addressing a detailed description of clinical, radiological, and histopathological features of this tumor.
CONCLUSION
Ependymal tumors mixed with ODG like clear cells constituted only 1.5% of all the CNS tumors presenting at our institute over past 11 years. A correct histopathological diagnosis aided with precise immunohistochemical stains and cytogenetic testing is critical for appropriate management and better outcome. In contrast to previous reports, we found majority of our patients (9 out of 11) had Grade III ependymal tumor with clear cells which may suggest a more aggressive disease on presentation, which may account for a poorer outcome in our cohort. However, our results were concordant with results reported from India, suggesting a possibility of more aggressive disease in this region.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
The authors would like to thank Dr. Khabir Ahmed for his assistance in writing the article.
References
1. Adamek D, Dec M, Sobol G, Urbanowicz B, Jaworski M. Giant cell ependymoma: A case report. Clin Neurol Neurosurg. 2008. 110: 176-81
2. Akutsu H, Shibata Y, Okazaki M, Hyodo A, Matsumura A. Intramedullary clear cell ependymoma in the cervical spinal cord: Case report. Neurosurgery. 2000. 47: 1434-7
3. Amatya VJ, Takeshima Y, Kaneko M, Nakano T, Yamaguchi S, Sugiyama K. Case of clear cell ependymoma of medulla oblongata: Clinicopathological and immunohistochemical study with literature review. Pathol Int. 2003. 53: 297-302
4. Brown DF, Chason DP, Schwartz LF, Coimbra CP, Rushing EJ. Supratentorial giant cell ependymoma: A case report. Mod Pathol. 1998. 11: 398-403
5. Cenacchi G, Giangaspero F, Cerasoli S, Manetto V, Martinelli GN. Ultrastructural characterization of oligodendroglial-like cells in central nervous system tumors. Ultrastruct Pathol. 1996. 20: 537-47
6. Deb P, Manu V, Pradeep H, Bhatoe HS. Intraparenchymal clear cell ependymoma. J Cytol. 2011. 28: 73-6
7. Dützmann S, Schatlo B, Lobrinus A, Murek M, Wostrack M, Weiss C. A multi-center retrospective analysis of treatment effects and quality of life in adult patients with cranial ependymomas. J Neurooncol. 2013. 114: 319-27
8. Fouladi M, Helton K, Dalton J, Gilger E, Gajjar A, Merchant T. Clear cell ependymoma: A clinicopathologic and radiographic analysis of 10 patients. Cancer. 2003. 98: 2232-44
9. Fourney DR, Siadati A, Bruner JM, Gokaslan ZL, Rhines LD. Giant cell ependymoma of the spinal cord. Case report and review of the literature. J Neurosurg. 2004. 100: 75-9
10. Fuller GN, Scheithauer BW. The 2007 revised World Health Organization (WHO) classification of tumours of the central nervous system: Newly codified entities. Brain Pathol. 2007. 17: 304-7
11. Hayashi K, Tamura M, Shimozuru T, Kasamo S, Hirahara K, Kadota K. Extra-axial ependymoma – Case report. Neurol Med Chir (Tokyo). 1994. 34: 295-9
12. Ho DM, Hsu CY, Wong TT, Chiang H. A clinicopathologic study of 81 patients with ependymomas and proposal of diagnostic criteria for anaplastic ependymoma. J Neurooncol. 2001. 54: 77-85
13. Jain D, Sharma MC, Arora R, Sarkar C, Suri V. Clear cell ependymoma: A mimicker of oligodendroglioma – Report of three cases. Neuropathology. 2008. 28: 366-71
14. Kakita A, Takahashi H, Fusejima T, Konno K, Nakazawa T, Aoki K. Clear cell variants of intracranial tumors: Meningioma and ependymoma. Noshuyo Byori. 1995. 12: 111-6
15. Katoh M, Satoh T, Nishiya M, Murata J, Ishii N, Saitoh H. Clear cell ependymoma of the fourth ventricle. Neuropathology. 2004. 24: 330-5
16. Kawano N, Yada K, Aihara M, Yagishita S. Oligodendroglioma-like cells (clear cells) in ependymoma. Acta Neuropathol. 1983. 62: 141-4
17. Kawano N, Yada K, Yagishita S. Clear cell ependymoma. A histological variant with diagnostic implications. Virchows Arch A Pathol Anat Histopathol. 1989. 415: 467-72
18. Kim YJ, Tsunoda S, Yokoyama K, Miyamoto K, Tamai M, Yamauchi M. Clear cell ependymoma with a lipidized component that developed in the thoracic spinal cord. Neurol Res. 2003. 25: 324-8
19. Kleihues P, Burger PC, Scheithauer BW. The new WHO classification of brain tumours. Brain Pathol. 1993. 3: 255-68
20. Kleihues P, Cavenee WK.editors. Pathology and Genetics of Tumours of the Nervous System. Lyon, France: International Agency for Research on Cancer; 2000. p.
21. Kurt E, Zheng PP, Hop WC, van der Weiden M, Bol M, van den Bent MJ. Identification of relevant prognostic histopathologic features in 69 intracranial ependymomas, excluding myxopapillary ependymomas and subependymomas. Cancer. 2006. 106: 388-95
22. Lee BH, Kwon JT, Park YS. Supratentorial clear cell ependymoma mimicking oligodendroglioma: Case report and review of the literature. J Korean Neurosurg Soc. 2011. 50: 240-3
23. Min KW, Scheithauer BW. Clear cell ependymoma: A mimic of oligodendroglioma: Clinicopathologic and ultrastructural considerations. Am J Surg Pathol. 1997. 21: 820-6
24. Nabors LB, Ammirati M, Bierman PJ, Brem H, Butowski N, Chamberlain MC. Central nervous system cancers. J Natl Compr Canc Netw. 2013. 11: 1114-51
25. Network NCC.editors. NCCN Clinical Practice Guidelines in Oncology: Central Nervous System Cancers. Ver. 2. Fort Washington, PA, USA: Network NCC; 2014; 2014. p.
26. Packer RR, Schiff D.editorsNeuro-oncology. Hoboken, NJ, USA: bJohn Wiley and Sons; 2012. p.
27. Parafeynikov V, Boaz JC, Bonnin JM. Clear-cell ependymoma: A report of intracortical tumor with significant desmoplasia. Neuropathology. 2008. 28: 165-70
28. Payet M, Conter C, Labrousse F, De Paula AM, Marabelle A, Branger DF. Clear cell ependymoma with trisomy 19 developing bone metastases. Childs Nerv Syst. 2012. 28: 739-42
29. Preusser M, Budka H, Rössler K, Hainfellner JA. OLIG2 is a useful immunohistochemical marker in differential diagnosis of clear cell primary CNS neoplasms. Histopathology. 2007. 50: 365-70
30. Rickert CH, Korshunov A, Paulus W. Chromosomal imbalances in clear cell ependymomas. Mod Pathol. 2006. 19: 958-62
31. Sato Y, Ochiai H, Yamakawa Y, Nabeshima K, Asada Y, Hayashi T. Brain surface ependymoma. Neuropathology. 2000. 20: 315-8
32. Seyithanoglu H, Guzey FK, Emel E, Alatas I, Acarbas A, Ozkan N. Clear cell ependymoma of the temporal lobe in a child: A case report. Pediatr Neurosurg. 2008. 44: 79-84
33. Suh JH, Hong SM, Lee IC. Clear cell ependymoma. Korean J Pathol. 1997. 31: 383-7
34. Teo C, Nakaji P, Symons P, Tobias V, Cohn R, Smee R. Ependymoma. Childs Nerv Syst. 2003. 19: 270-85

