

- Northwestern University Feinberg School of Medicine, Chicago, Illinois, USA
- Medical School for International Health, Beersheba, Israel
- Department of Biochemistry and Molecular Genetics, Northwestern University Feinberg School of Medicine, Chicago, Illinois, USA
- Department of Neurological Surgery, Northwestern University Feinberg School of Medicine, Chicago, Illinois, USA
Correspondence Address:
Michel Kliot
Department of Neurological Surgery, Northwestern University Feinberg School of Medicine, Chicago, Illinois, USA
DOI:10.4103/2152-7806.194058
Copyright: © 2016 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Krish Suresh, Tamara Kliot, Andrea Piunti, Michel Kliot. Epigenetic mechanisms drive the progression of neurofibromas to malignant peripheral nerve sheath tumors. 14-Nov-2016;7:
How to cite this URL: Krish Suresh, Tamara Kliot, Andrea Piunti, Michel Kliot. Epigenetic mechanisms drive the progression of neurofibromas to malignant peripheral nerve sheath tumors. 14-Nov-2016;7:. Available from: http://surgicalneurologyint.com/surgicalint_articles/epigenetic-mechanisms-drive-progression-neurofibromas-malignant-peripheral-nerve-sheath-tumors/
Date of Submission
18-Mar-2016
Date of Acceptance
01-Aug-2016
Date of Web Publication
14-Nov-2016
Abstract
Thinking Outside the Box:The polycomb repressive complex 2 (PRC2) is a histone methyltransferase complex known to repress gene expression. There is a large body of experimental evidence that supports its role in promoting tumorigenicity by suppressing tumor suppressor genes. Here, we discuss the surprising findings that, in neurofibromas, it may have a completely different role as a tumor suppressor; mutations of PRC2 lead to conversion of benign neurofibromas into malignant peripheral nerve sheath tumors (MPNSTs) by de-repressing and thereby activating genes driving cell growth and development. These findings have potentially powerful clinical applications in both diagnosing and treating MPNSTs.
Hypothesis:PRC2 loss drives malignant transformation of neurofibromas.
Keywords: Epigenetics, MPNST, neurofibroma, PRC2
INTRODUCTION
Neurofibromas are benign tumors of the peripheral nerve sheath. Their origin is heterogeneous and includes schwann cells, neurons, fibroblasts, perineurial cells,[
NF1 patients develop neurofibromas following a second hit mutation to the NF1 gene; this second hit is essential for neurofibroma development, showing that neurofibromin is a tumor suppressor.[
Once a patient has developed a neurofibroma, its progression over time is variable. Many stop growing, some continue to grow, and very few may develop into malignant peripheral nerve sheath tumors (MPNSTs).[
The neurofibromas that do develop into MPNSTs have poor prognoses. MPNSTs are classified as sarcomas (malignant mesenchymal tumors) and have 5-year survival rates as low as 35%.[
There are several genetic factors that drive the progression of neurofibromas to malignancy. Many studies have shown that MPNSTs harbor mutations in cell cycle regulators including CDKN2A/B and p53; these mutations are rarely found in benign neurofibromas, demonstrating that loss of these regulators is a key event in the development of malignancy.[
Genetic factors are also implicated in the growth arrest observed in many neurofibromas. One study showed that loss of neurofibromin initially triggers the activation of Ras, however, this subsequently triggers a global negative feedback signaling program resulting in “oncogene induced senescence.” This program involves many genes (Sprouty, HDM2, FOXO) that converge on the p53 and Rb pathways to arrest the cell cycle [
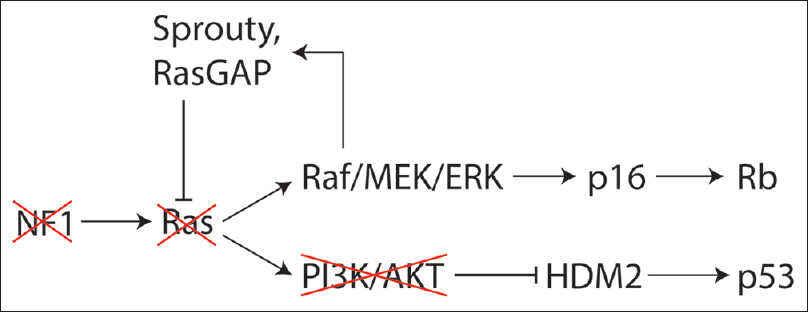
EPIGENETICS OF MALIGNANT PERIPHERAL NERVE SHEATH TUMORS
Epigenetic mechanisms have also been shown to play a significant role in the progression of neurofibromas to MPNSTs. These revolve around the polycomb repressive complex 2 (PRC2). PRC2 is a protein complex that methylates lysine 27 on the tail of histone 3 (H3K27), resulting in transcriptional repression. PRC2 is generally regarded as an oncogene as it is up-regulated in many cancers,[
In one study, 70% of NF-1 associated MPNSTs (and over 90% of radiotherapy-associated and sporadic MPNSTs) harbored loss-of-function mutations in PRC2.[
PRC2 mutations may also further activate the Ras pathway, already amplified by the loss of neurofibromin [
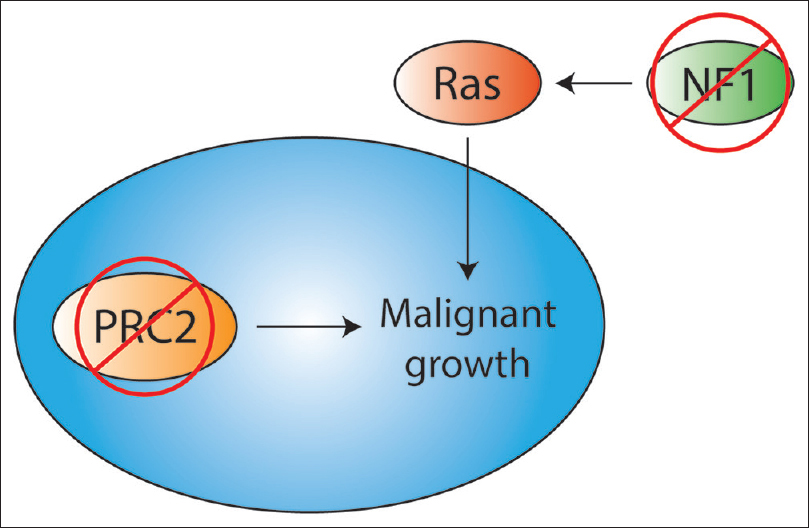
PRC2 mutations are not sufficient for malignancy alone; rather they cooperate with mutations in cell cycle regulators. Indeed, the MPNSTs in the previously described studies harbored CDKN2A mutations in addition to PRC2 mutations.[
Another intriguing relationship between PRC2 and NF1 is that the gene for SUZ12, a component of the PRC2 complex, is 560 kb telomeric to the NF1 gene on chromosome 17q, and they are often codeleted in NF1.[
While most studies in this area show that PRC2 is tumor suppressive in MPNSTs, there is one study worth noting that takes the opposite view. This study showed that EZH2, a PRC2 component, is up-regulated in MPNSTs. They propose that, in this context, EZH2 promotes a nuclear transport receptor that drives MPNST development.[
There are relatively few studies on the epigenetic mechanisms driving growth arrest in neurofibromas, but PRC2 may be involved. One study found that in benign neurofibromas, JMJD3—a protein that demethylates H3K27, thus activating genes previously repressed by PRC2—was up-regulated. Consequently, the Ink4a/Arf locus was activated, inducing senescence. Accordingly, transgenic mice made to develop neurofibromas showed reduced levels of H3K27me3 at the Ink4a/Arf locus in the benign tumors.[
These findings have paved the way for more detailed models of how MPNSTs may develop from neurofibromas. One model posits that, if the first or second hit to the NF1 gene is a deletion that encompasses SUZ12, then a third hit to the remaining SUZ12 gene drives MPNST development.[
AVENUES FOR DIAGNOSIS AND TREATMENT
The role of PRC2 in the development of MPNSTs presents new avenues for diagnosis and treatment. One diagnostic method makes use of the mark that PRC2 leaves—tri-methylated H3K27 (H3K27me3). A recent study showed that loss of H3K27me3 could be a highly specific diagnostic test for distinguishing MPNSTs from histological mimics. Furthermore, high-grade MPNSTs showed more complete H3K27me3 loss than lower-grade tumors, hence, this test may be used to grade MPNSTs as well.[
It follows that a therapeutic avenue is the reintroduction of wild type PRC2 in MPNSTs, in order to re-methylate the appropriate genes and suppress their transcription. Indeed, this strategy was shown to partially restore the histone methylation signature of the genome and decrease cell growth.[
Another potential therapy that rescues the effects of PRC2 loss involves bromodomain inhibitors such as JQ1. When PRC2 is lost and H3K27 methylation cannot be maintained, H3K27 becomes acetylated (H3K27Ac). H3K27Ac recruits bromodomain proteins, which play a role in transcriptional activation.[
Lastly, the opposing study noted in the previous section was followed up with another by the same authors exploring the therapeutic implications. They showed that EZH2 inhibitors effectively down-regulated a nuclear transport protein, leading to reduced proliferation and eventual apoptosis of MPNST cells.[
NEXT STEPS
Further study of the epigenetic mechanisms regulating the development and progression of neurofibromas could lead to improved diagnosis and treatment of these tumors. In addition to treating MPNSTs, it will be interesting to see if these strategies could arrest the progression of neurofibromas into MPNSTs in the first place. Many neurofibromas spontaneously stop growing, and perhaps epigenetic mechanisms drive this arrest. PRC2 seems a good place to search for answers to these questions.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Baude A, Lindroth AM, Plass C. PRC2 loss amplifies Ras signaling in cancer. Nat Genet. 2014. 46: 1154-5
2. Beert E, Brems H, Daniels B, De Wever I, Van Calenbergh F, Schoenaers J. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer. 2011. 50: 1021-32
3. Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003. 22: 5323-35
4. Cichowski K, Shih TS, Schmitt E, Santiago S, Reilly K, McLaughlin ME. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999. 286: 2172-6
5. Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell. 2006. 10: 459-72
6. De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014. 514: 247-51
7. De Raedt T, Brems H, Wolkenstein P, Vidaud D, Pilotti S, Perrone F. Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet. 2003. 72: 1288-92
8. Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Medical Genet. 2002. 39: 311-4
9. Gomez-Sanchez JA, Gomis-Coloma C, Morenilla-Palao C, Peiro G, Serra E, Serrano M. Epigenetic induction of the Ink4a/Arf locus prevents Schwann cell overproliferation during nerve regeneration and after tumorigenic challenge. Brain. 2013. 136: 2262-78
10. Holtkamp N, Mautner VF, Friedrich RE, Harder A, Hartmann C, Theallier-Janko A. Differentially expressed genes in neurofibromatosis 1-associated neurofibromas and malignant peripheral nerve sheath tumors. Acta Neuropathol. 2004. 107: 159-68
11. Kliot T, Ince Y, Tihan T, Wilson M, Kliot M. To grow or not to grow, That is the question. Surg Neurol Int. 2013. 4: S407-10
12. Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014. 46: 1227-32
13. Maertens O, Cichowski K. An expanding role for RAS GTPase activating proteins (RAS GAPs) in cancer. Adv Biol Regul. 2014. 55: 1-14
14. Pasini D, Malatesta M, Jung HR, Walfridsson J, Willer A, Olsson L. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res. 2010. 38: 4958-69
15. Perrone F, Tabano S, Colombo F, Dagrada G, Birindelli S, Gronchi A. p15INK4b, p14ARF, and p16INK4a inactivation in sporadic and neurofibromatosis type 1-related malignant peripheral nerve sheath tumors. Clin Cancer Res. 2003. 9: 4132-8
16. Piunti A, Rossi A, Cerutti A, Albert M, Jammula S, Scelfo A. Polycomb proteins control proliferation and transformation independently of cell cycle checkpoints by regulating DNA replication. Nat Commun. 2014. 5: 3649-
17. Rahrmann EP, Watson AL, Keng VW, Choi K, Moriarity BS, Beckmann DA. Forward genetic screen for malignant peripheral nerve sheath tumor formation identifies new genes and pathways driving tumorigenesis. Nat Genet. 2013. 45: 756-66
18. Schaefer IM, Fletcher CD, Hornick JL. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod Pathol. 2016. 29: 4-13
19. Thomas L, Mautner VF, Cooper DN, Upadhyaya M. Molecular heterogeneity in malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1. Hum Genomics. 2012. 6: 18-
20. Tucker T, Friedman JM, Friedrich RE, Wenzel R, Funsterer C, Mautner VF. Longitudinal study of neurofibromatosis 1 associated plexiform neurofibromas. J Med Genet. 2009. 46: 81-5
21. Tucker T, Riccardi VM, Sutcliffe M, Vielkind J, Wechsler J, Wolkenstein P. Different patterns of mast cells distinguish diffuse from encapsulated neurofibromas in patients with neurofibromatosis 1. J Histochem Cytochem. 2011. 59: 584-90
22. Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002. 419: 624-9
23. Zhang M, Wang Y, Jones S, Sausen M, McMahon K, Sharma R. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet. 2014. 46: 1170-2
24. Zhang P, Garnett J, Creighton CJ, Al Sannaa GA, Igram DR, Lazar A. EZH2-miR-30d-KPNB1 pathway regulates malignant peripheral nerve sheath tumour cell survival and tumourigenesis. J Pathol. 2014. 232: 308-18
25. Zhang P, Yang X, Ma X, Ingram DR, Lazar AJ, Torres KE. Antitumor effects of pharmacological EZH2 inhibition on malignant peripheral nerve sheath tumor through the miR-30a and KPNB1 pathway. Mol Cancer. 2015. 14: 55-
26. Zhu Y, Parada LF. Neurofibromin, a tumor suppressor in the nervous system. Exp Cell Res. 2001. 264: 19-28
27. Zou C, Smith KD, Liu J, Lahat G, Myers S, Wang WL. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg. 2009. 249: 1014-22

