

- Neurosurgery Department, DFV Neuro, São Paulo, Brazil
- Hospital Sírio Libanês, São Paulo, Brazil
- Hospital Alemão Oswaldo Cruz, São Paulo, Brazil
Correspondence Address:
Matheus Fernandes de de Oliveira
Neurosurgery Department, DFV Neuro, São Paulo, Brazil
Hospital Sírio Libanês, São Paulo, Brazil
Hospital Alemão Oswaldo Cruz, São Paulo, Brazil
DOI:10.4103/sni.sni_319_17
Copyright: © 2018 Surgical Neurology International This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.How to cite this article: Eduardo de Arnaldo Silva Vellutini, Pedro Henrique Petit Becker, Luis Felipe Godoy, Nicolau Faria Correia Guerreiro, Romulo Loss Mattedi, Matheus Fernandes de de Oliveira. Epithelioid pituicytoma: An unusual case report. 24-Jul-2018;9:145
How to cite this URL: Eduardo de Arnaldo Silva Vellutini, Pedro Henrique Petit Becker, Luis Felipe Godoy, Nicolau Faria Correia Guerreiro, Romulo Loss Mattedi, Matheus Fernandes de de Oliveira. Epithelioid pituicytoma: An unusual case report. 24-Jul-2018;9:145. Available from: http://surgicalneurologyint.com/surgicalint-articles/epithelioid-pituicytoma-an-unusual-case-report/
Date of Submission
24-Aug-2017
Date of Acceptance
14-Jun-2018
Date of Web Publication
24-Jul-2018
Abstract
Background:Pituicytomas are considered World Health Organization Grade I malignancies. Until September 2017, a total of 81 cases of pituicytomas were diagnosed and described in literature. We present such a case in which histopathology shows an epithelioid pattern, a rare variant of pituicytoma. As far as we know, this is only the second such case described in the literature.
Case Description:A 61-year-old male patient presented with complaints of progressive decrease in visual acuity for about 7 months, worse on the left side. Laboratory and endocrinological investigation returned normal values. Magnetic resonance imaging revealed a mixed solid-cystic lesion, measuring about 3.1 × 2.2 × 2.9 cm. The lesion presented with intermediate signal intensity in T1 and T2 sequences and showed avid postcontrast enhancement. The patient underwent resection through a left pterional approach. Pathology revealed a glial neoplasm with an epithelioid pattern and moderate cellularity with rounded-elongated cell nuclei and with a broad eosinophilic cytoplasm. Absence of cellular pleomorphism, any mitotic figures, or areas of necrosis was noted.
Conclusion:The epithelioid variant of pituicytomas differs from the commonly encountered forms of this tumor which typically present in a fascicular pattern. Microsurgical resection is the treatment of choice. However, in many cases, subtotal resection was performed because of a considerable risk for neurovascular injuries.
Keywords: Brain neoplasm, pathology, pituicytoma treatment
INTRODUCTION
Primary non-adenomatous tumors of the pituitary gland are comparatively rare entities and have been classified according to the 2007 and 2016 World Health Organization (WHO) into three different histological subtypes: tumors of pituicytes, tumors of granular cells, and oncocytomas of spindle cells.[
Histologically, these lesions are usually solid and made up by spindle-shaped cells in fascicular arrangements.[
CASE DESCRIPTION
History
A 61-year-old male patient complained of progressive decrease in visual acuity over 7 months before admission, worse on the left side. He also reported increased urine output for 3 months. His medical history included a left lung lobectomy for lung cancer 25 years earlier. Neurological examination did not show any other abnormalities. Laboratory and endocrinological investigation were normal.
Radiographic features
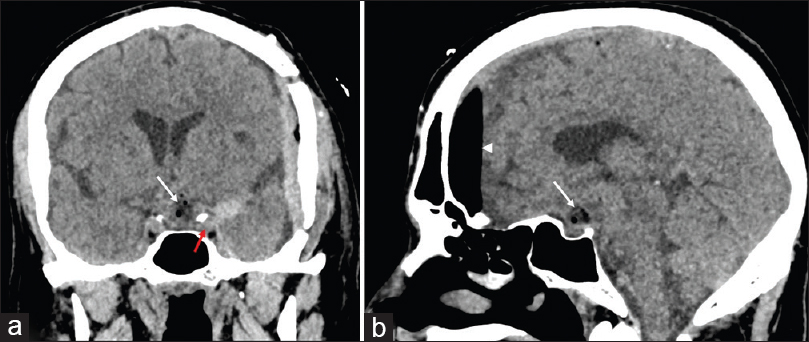
Magnetic resonance imaging (MRI) revealed a mixed solid-cystic lesion measuring 3.1 × 2.2 × 2.9 cm and appearing with intermediate signal on T1 and T2 sequences and with avid postcontrast enhancement after application of i/v gadolinium. The lesion was centered in the suprasellar cistern at the expected site of pituitary stalk and compressed and displaced the optic chiasm. The tumor extended superiorly toward the floor of the third ventricle, compressing the hypothalamus bilaterally, without an apparent discernable tissue plane separating it from adjacent structures [
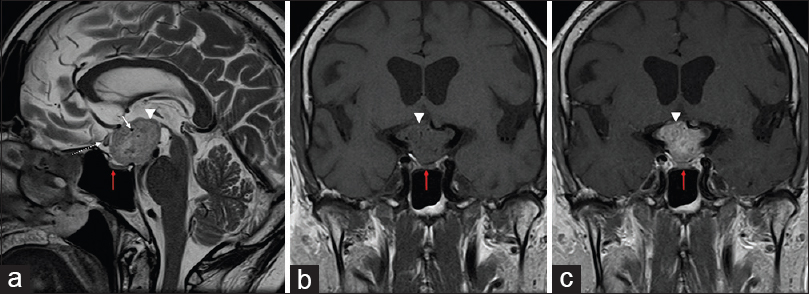
Figure 1
Magnetic resonance (MR) images in sagittal T2 (a), T1 coronal (b), and T1 coronal after contrast (c) reveal a lesion in suprasellar cistern. It is characterized by heterogeneous hyperintensity in T2, with gross enhancement by gadolinium (arrow heads). Vascular structures are seen in the interior of lesion, with flow-voids (white arrows). It compresses and pushes anteriorly the optic chiasm, changing its sign (descontinuous arrow). Adenohypophysis is apart from the lesion (red arrow)
Posteriorly, the lesion displayed intimate contact with the pituitary stalk and extended into the interpeduncular cistern, displacing the mesencephalon posterior-inferiorly. On T2 and FLAIR sequences, there was increased signal intensity along the pathway of the optic tract and in the hypothalamus which was interpreted as edema. The usual T1-signal increase of the neuropituitary was not visualized and the appearance of adenohypophysis was normal.
Surgical management
The patient underwent open microsurgery through a left pterional approach. The lesion was found to be firm and could not be aspirated by suction-irrigation and was rather hypervascular, leading to significant intraoperative blood loss. The tumor appeared to have originated from the pituitary stalk which was infiltrated. The chiasm was elevated and the left optic tract was displaced laterally by the tumor. A good cleavage plane could be identified, and after subtotal removal (limited by hypothalamic infiltration and involvement of perforating arteries of the posterior communicating and the posterior cerebral artery), we achieved a substantial decompression of the optic pathway. The postoperatively period was uneventful, except some transient diabetes insipidus which resolved spontaneously by the fourth postoperative day [
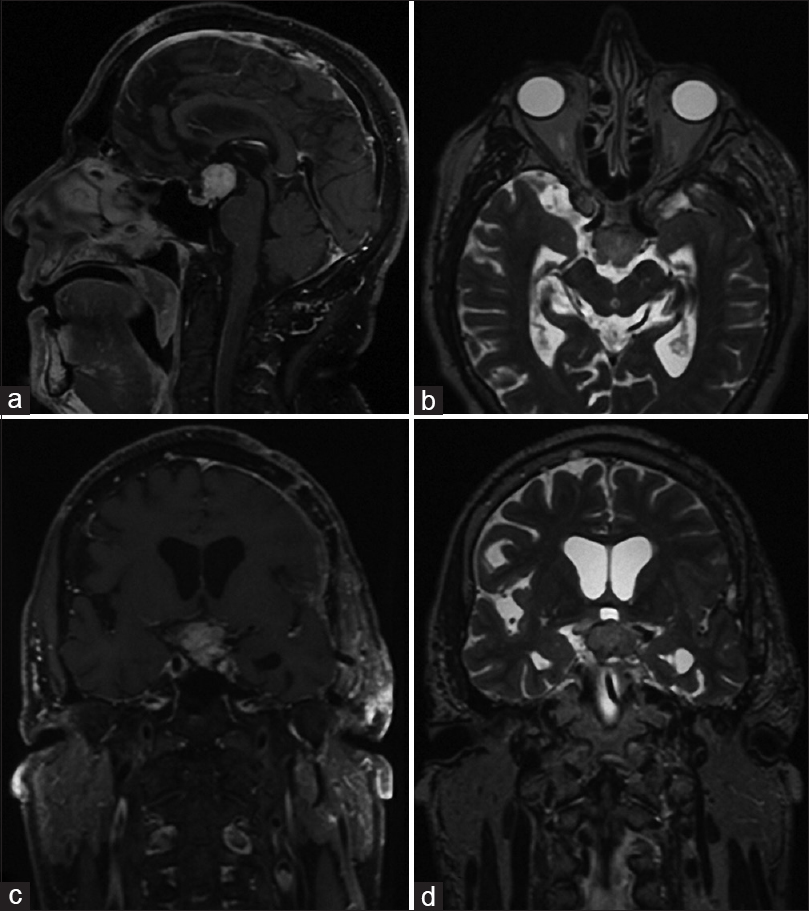
Pathology
Macroscopic pathological examination revealed a lesion of whitish-brown discoloration with an elastic consistency. Histopathology revealed a glial neoplasm with an epithelioid pattern. The specimen showed moderate cellularity with sometimes rounded or elongated nuclei and a broad eosinophilic cytoplasm. Absence of cellular pleomorphism, mitotic figures, and any areas of necrosis was noted. The immunohistochemical staining profile showed positivity for GFAP, S-100, Vimentin, TTF-1, ATRX, and BCL-2. Staining was negative for EMA, Synaptophysin, P53, HDI-1, NEUN, and CK8/18; The cell proliferation index (estimated by Ki-67) was low at about 1% [
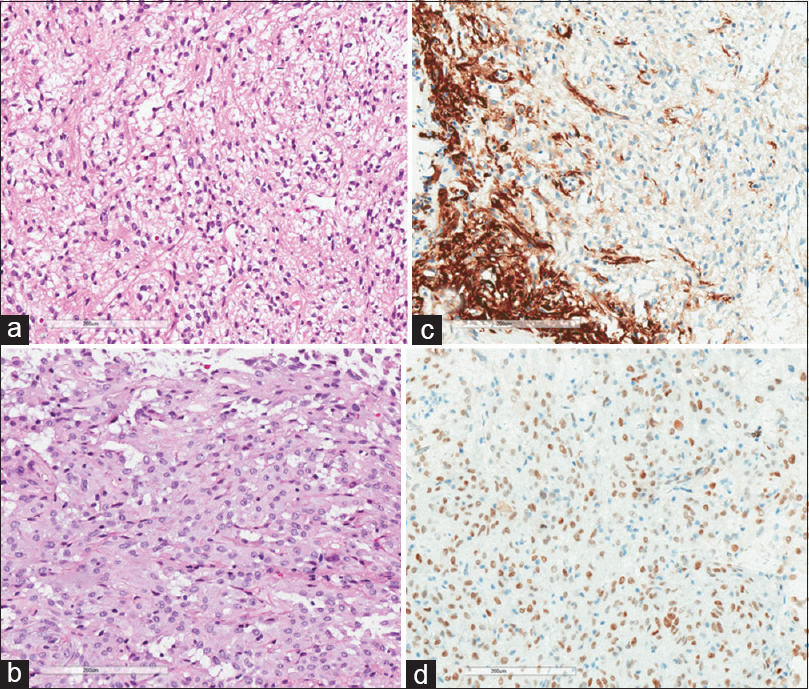
Figure 4
Histology image. (a) Hematoxylin–eosin ×200 view; fusiform cells. (b) Hematoxylin–eosin ×200; revealing epithelioid pattern with moderate cellularity, sometimes rounded nuclei, sometimes elongated, and broad eosinophilic cytoplasm. (c) Positive staining for GFAP. (d) Positive staining for TTF-1
DISCUSSION
The 2007 WHO classification of CNS pituitary neoplasms was confirmed in the 2016 update and distinguishes three separate non-adenomatous pituitary tumour entities with distinct morphological, immunohistochemical, and ultrastructural features: granular cell tumors, spindle cell oncocytomas, and pituicytomas. The fact that these neoplasms all share the characteristic feature of nuclear expression of thyroid transcription factor-1 (TTF-1) raises the possibility that they may actually constitute a spectrum of a single nosological entity which is derived from posterior pituitary glial cells, the pituicytes.[
Pituicytes are specialized cells of ependymal origin found in neurohypophysis and constitute five different cell types (larger cells, dark cells, oncocytic cells, granular cells, and ependymal cells). It is believed that most pituicytomas originate from the first two types listed.
The epithelioid variant of pituicytomas is a recently discovered entity and has been described only once thus far by Ellis et al.[
Symptoms of pituicytomas are similar to other sellar lesions and are due to mass effect resulting in headaches, visual deficits, and sexual dysfunction. They may also cause fatigue, hypopituitarism, signs of increased serum prolactin levels, and less frequently diabetes insipidus. Due to its slow growth rate of pituicytomas, symptoms appear late but may present an acutely secondary to tumor apoplexy.[
MR is the diagnostic modality of choice, showing usually a suprasellar solid lesion with iso- or hyperintense signal on T2 sequences, isointense signal on T1 sequences, and homogeneous contrast uptake after gadolinium application. Another important feature is the unremarkable appearance of the pituitary gland itself. Radiographic differential diagnoses include cordoid glioma, papillary craniopharyngioma, and granular cell tumor of the pituitary stalk.[
Macroscopically, most pituicytomas are solid tumors of firm consistency, well delineated, and without any tendency to infiltrate adjacent structures. Microscopically, these lesions are formed by bipolar spindle cells, with oval nuclei, eosinophilic cytoplasm without granules, with minimal cellular atypia, and without mitosis, but with Rosenthal fibers. The cytoarchitecture shows an arrangement of cells in fascicles.[
Pituicyomas stain positive for Vimentin, Protein S-100, and GFAP, suggesting a glial origin. Positivity on TFF-1 stain is also characteristic.[
The exceedingly rare epithelioid variant described here differs from usual pituicytomas due to its cytoarchitecture, which instead of presenting with a fascicular pattern shows an epithelioid pattern.[
Surgery for microscopic resection is the treatment of choice. However, in many cases, only subtotal resection can be achieved because of significant intraoperative bleeding and the risk of injury to adjacent neurological structures due to tumor adhesions.[
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Covington MF, Chin SS, Osborn AG. Pituicytoma, spindle cell oncocytoma, and granular cell tumor: Clarification and meta-analysis of the world literature since 1893. AJNR Am J Neuroradiol. 2011. 32: 2067-72
2. Ellis JA, Tsankova NM, D'Amico R, Ausiello JC, Canoll P, Rosenblum MK. Epithelioid pituicytoma. World Neurosurg. 2012. 78: 191.E1-7
3. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathology. 2007. 114: 97-109
4. Sahm F, Reuss DE, Giannini C. WHO 2016 Classification: Changes and advancements in the diagnosis of miscellaneous primary CNS tumours. Neuropathol Appl Neurobiol. 2018. 44: 163-71
5. Tian Y, Yue S, Jia G, Zhang Y. Childhood giant pituicytoma: A report and review of the literature. Clin Neurol Neurosurg. 2013. 115: 1943-50
6. Ulm AJ, Yachnis AT, Brat DJ, Rhoton AL. Pituicytoma: Report of two cases and clues regarding histogenesis. Neurosurgery. 2004. 54: 753-8
7. Wang J, Liu Z, Du J, Cui Y, Fang J, Xu L. The clinicopathological features of pituicytoma and the differential diagnosis of sellar glioma. Neuropathology. 2016. 36: 432-40
8. Wolfe SQ, Bruce J, Morcos JJ. Pituicytoma: Case report. Neurosurgery. 2008. 63: E173-4
9. Yang X, Liu X, Li W, Chen D. Pituicytoma: A report of three cases and literature review. Oncol Lett. 2016. 12: 3417-22
10. Zygourakis CC, Rolston JD, Lee HS, Partow C, Kunwar S, Aghi MK. Pituicytomas and spindle cell oncocytomas: Modern case series from the University of California, San Francisco. Pituitary. 2015. 18: 150-8

