

- Department of Neurosurgery, Medicine Maastricht University Medical Center, Maastricht, Limburg, Netherlands.
- Department of Pathology, Medicine Maastricht University Medical Center, Maastricht, Limburg, Netherlands.
- Department of Medical Oncology Medicine Maastricht University Medical Center, Maastricht, Limburg, Netherlands.
- Department of Neurology, Medicine Maastricht University Medical Center, Maastricht, Limburg, Netherlands.
- Department of Radiotherapy, Maastro Clinic, Maastricht, Limburg, Netherlands.
- Department of Radiology and Nuclear, Medicine Maastricht University Medical Center, Maastricht, Limburg, Netherlands.
Correspondence Address:
Rutger J. Slegers, Department of Neurosurgery, Medicine Maastricht University Medical Center, Maastricht, Limburg, Netherlands.
DOI:10.25259/SNI_1153_2021
Copyright: © 2022 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Rutger J. Slegers1, Jan Beckervordersandforth2, Ann Hoeben3, Govert Hoogland1, Martijn P. G. Broen4, Monique Anten4, Jim T. A. Dings1, Piet van den Ende5, Wouter J. P. Henneman6, Olaf E. M. G. Schijns1. From a dysembryoplastic neuroepithelial tumor to a glioblastoma multiforme: Pitfalls of initial diagnosis on biopsy material, a case report. 11-Feb-2022;13:43
How to cite this URL: Rutger J. Slegers1, Jan Beckervordersandforth2, Ann Hoeben3, Govert Hoogland1, Martijn P. G. Broen4, Monique Anten4, Jim T. A. Dings1, Piet van den Ende5, Wouter J. P. Henneman6, Olaf E. M. G. Schijns1. From a dysembryoplastic neuroepithelial tumor to a glioblastoma multiforme: Pitfalls of initial diagnosis on biopsy material, a case report. 11-Feb-2022;13:43. Available from: https://surgicalneurologyint.com/surgicalint-articles/11387/
Date of Submission
18-Nov-2021
Date of Acceptance
15-Jan-2022
Date of Web Publication
11-Feb-2022
Abstract
Background: Ganglioglioma (GG) and dysembryoplastic neuroepithelial tumor (DNET) belong to the group of low-grade epilepsy-associated tumors (LEAT) and are the most prevalent tumor types found in patients undergoing epilepsy surgery. Histopathological differentiation between GG and DNET can be difficult on biopsies due to limited tumor tissue.
Case Description: Here, we present a rare case where a low-grade tumor was initially classified as DNET, based on biopsy findings and unfortunately dedifferentiated within 10 years into a glioblastoma multiforme. After gross total resection, the initial tumor was reclassified as GG.
Conclusion: This case illustrates the diagnostic challenges of LEAT, especially on biopsy material. Therefore, we advocate to counsel for complete resection and histopathological diagnosis utilizing tumor markers to confirm the nature of the tumor and to advice type of follow-up and eventual concurrent treatment.
Keywords: Dysembryoplastic neuroepithelial tumor, Ganglioglioma, Glioblastoma, Low-grade epilepsy-associated tumor, Temporal lobe epilepsy
INTRODUCTION
Ganglioglioma (GG) comprises 0.93% of all primary central nervous system tumors, is frequently classified as WHO Grade I neoplasms, and belongs to the Low-grade epilepsy-associated tumor (LEAT) spectrum.[
CASE DESCRIPTION
A 38-year-old Caucasian woman was admitted to our hospital in 2006 for focal epileptic seizures. Seizures consisted of paroxysmal contractions of the right shoulder without loss of consciousness at a frequency of 3 times/week. MRI-scan showed a non-enhancing, left-sided temporo-parieto-occipital tumor [
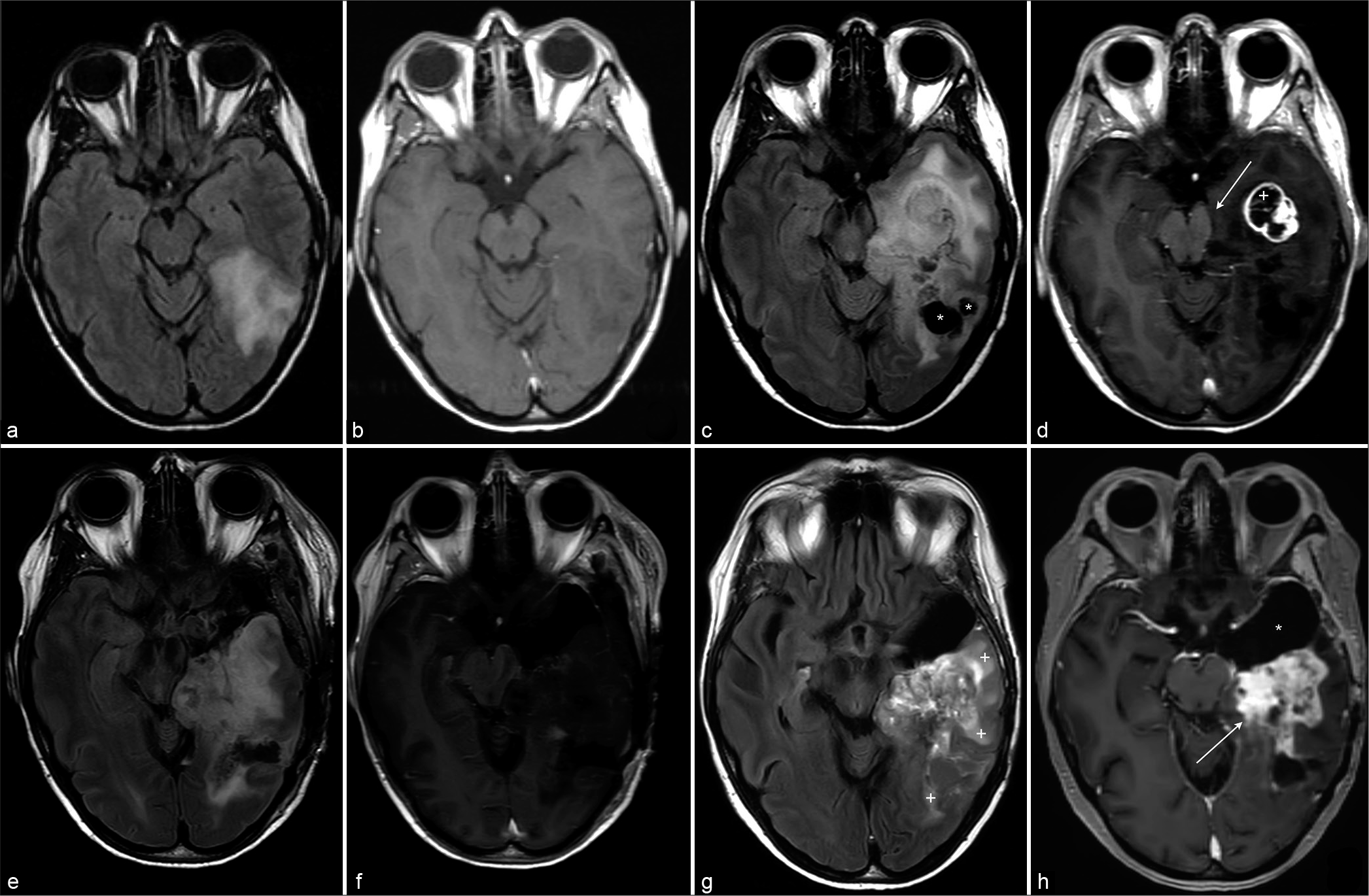
Figure 1:
(a) Transverse FLAIR and (b) contrast-enhanced T1 at presentation show a non-enhancing T2-hyperintense mass in the left temporal lobe. These findings favor a low-grade tumor but are otherwise non-specific. (c) Transverse FLAIR and (d) contrast-enhanced T1 10 years after first presentation. There is evident progression of the mass, with cystic components (*) and a rim-enhancing component (+) in the anterior temporal lobe. Increase of transtentorial herniation with mass-effect on the brainstem (arrow). (e) Transverse Flair and (f) Contrast enhanced T1 Postoperative within 72 h shows slight residual pathological contrast enhancement at the resection margins, suggesting minimal rest tumor. (g) Transverse FLAIR and (h) contrast enhanced T1 13 years after first presentation. Despite anterior debulking (*marks the postoperative defect), there is evident progression of enhancing tumor (arrow) and surrounding edema (+).
For 8 years the seizures remained under control and the routine follow-up MRI-scan showed no signs of tumor progression. Shortly thereafter, the patient was admitted with agitation, dysphasia, and visual hallucinations in the right visual field. At admission, consciousness was intact and dysphasia, apraxia and a right-sided facial paresis was observed and levetiracetam was increased to 750 mg twice daily. An MRI-scan showed tumor growth, from 5.0 × 4.0 cm in 2006 to 7.6 × 5.8 cm in 2014. The tumor had solid and cystic components and did not enhance after gadolinium contrast. Mass effect was seen with slight displacement of the mesencephalon. The dysphasia and visual symptoms were interpreted as a combination of transient tumor (seizure) attacks and a mass effect on eloquent temporal neocortex and the optic radiation. The patient was scheduled for an awake craniotomy (Penfield procedure) but declined surgical treatment in the end. Ten years after the first admission, the MRI-scan showed further tumor progression with increased compression of the mesencephalon [
Control MRI-scan, 6-months after surgery showed decreased gliotic changes but also progression of both enhancing and non-enhancing nodular components with regions of increased perfusion ratios, indicative of further progression of dedifferentiated tumor components. One month after this MRI-scan, the patient presented on the ER with an acute visual aura, numbness in the right half of the face, weakness of the right arm, dysarthria, and dysphasia, and interpreted as postictal symptoms. Physical examination showed a latent paresis of the right arm and no further deficits. Subsequent MRI-scan showed further tumor progression [
Histopathology
The biopsy showed brain tissue with increased cellularity, a variable distribution, and a remarkably perivascular pattern, suggestive for a low-grade primary brain tumor [
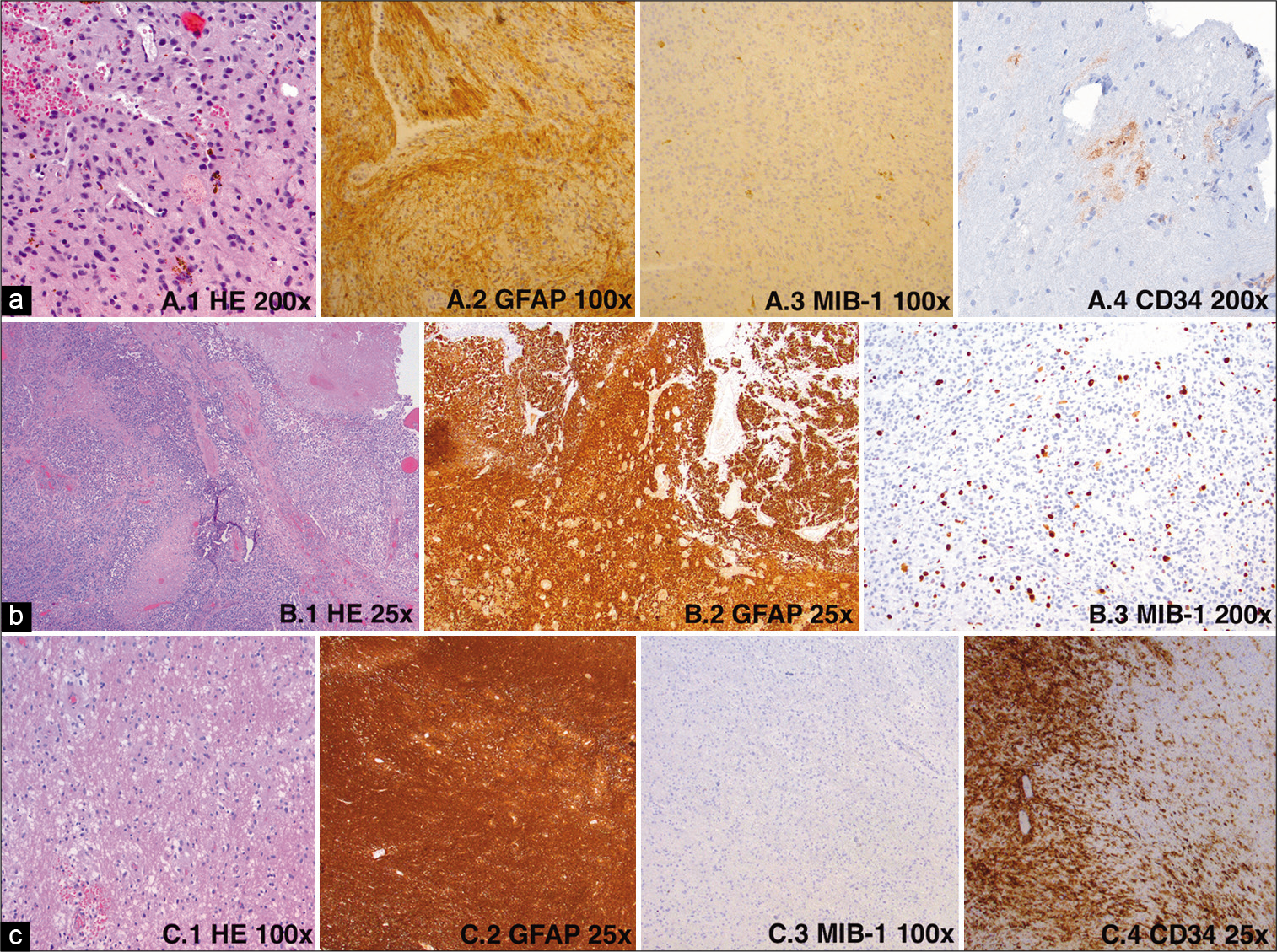
Figure 2:
(a) First tumor biopsy HE-staining (1), GFAP staining (2), proliferation marker Mib1 (3) and later performed staining for CD34 (4), (b) high grade recurrence as GBM (resection) HE-staining (1), GFAP staining (2), proliferation marker Mib1 (3), (c) Resection of hippocampus (place of prior biopsy) with tumor in HE-staining (1), GFAP staining (2), staining for the proliferation marker Mib1 (3) and CD34 (4).
DISCUSSION
This case report describes the complexity of the histopathological differentiation between tumors in the LEAT-spectrum, especially under the circumstances of limited tissue after tumor biopsy without resection. Besides, this report describes the urge of a correct diagnosis for the purpose of a meaningful prognostic patient counseling.
GG was first described by Perkins in 1926[
The advent and application of different diagnostic tumor markers, such as CD34, have led to an increased recognition of these markers in the tumors of the LEAT spectrum. CD34 was first described in 1999 and in the LEAT spectrum 54/73 (74%) GG’s and 4/23 (17%) DNET’s showed immunoreactivity for this novel marker.[
The importance of differentiating between these LEAT-spectrum tumors is the difference in prognosis between the tumor groups and the eventual postoperative follow-up. Due to the presence of a glial component in GG, these tumors can dedifferentiate into a high-grade glioma.[
In the presented case, the main difficulty was the amount of available tissue at initial diagnosis obtained after a biopsy. It is speculated that, at first presentation, histomorphological analysis of a larger tissue volume obtained after resection, together with the use of specific markers as mentioned above would have led to the correct GG diagnosis. In case, the amount of available tissue is sparse and specific tumor markers are absent, discriminating within the LEAT spectrum presents significant difficulties. As the current case illustrates, initial correct diagnosis is critically important to estimate the overall and progression-free survival and counsel the patient for prognosis and eventual treatment. In conclusion, in future cases of a suspected LEAT tumor the difficulty and chance of correct diagnosis and its implications using biopsy versus resection should be discussed. We advocate to counsel the patient in these cases for complete resection and the standard use of the above-mentioned tumor markers to confirm the exact nature of the tumor. This is necessary to advise the type of follow-up, decide on eventual concurrent treatment and to counsel the patient, when requested, with an estimation of the prognosis quoad vitam. In case of an unexpected tumor progression in a biopsy-diagnosed DNET, it is advisable to counsel the patient for a second look and gross-total resection.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
The authors thank the patient, family, and all the people who participated in this case report, hoping to make a contribution to the research. Informed consent was obtained from all individual participants included in the study.
References
1. Blumcke I, Coras R, Wefers AK, Capper D, Aronica E, Becker A. Review: Challenges in the histopathological classification of ganglioglioma and DNT: Microscopic agreement studies and a preliminary genotype-phenotype analysis. Neuropathol Appl Neurobiol. 2019. 45: 95-107
2. Blumcke I, Giencke K, Wardelmann E, Beyenburg S, Kral T, Sarioglu N. The CD34 epitope is expressed in neoplastic and malformative lesions associated with chronic, focal epilepsies. Acta Neuropathol. 1999. 97: 481-90
3. Blumcke I, Spreafico R, Haaker G, Coras R, Kobow K, Bien CG. Histopathological findings in brain tissue obtained during epilepsy surgery. N Engl J Med. 2017. 377: 1648-56
4. Courville CB. Ganglioglioma: Tumor of the central nervous system; review of the literature and report of two cases. Arch Neurol Psychiatr. 1930. 24: 439-91
5. Darlix A, Zouaoui S, Rigau V, Bessaoud F, Figarella-Branger D, Mathieu-Daude H. Epidemiology for primary brain tumors: A nationwide population-based study. J Neurooncol. 2017. 131: 525-46
6. Daumas-Duport C, Scheithauer BW, Chodkiewicz JP, Laws ER, Vedrenne C. Dysembryoplastic neuroepithelial tumor: A surgically curable tumor of young patients with intractable partial seizures. Report of thirty-nine cases. Neurosurgery. 1988. 23: 545-56
7. Heiland DH, Staszewski O, Hirsch M, Masalha W, Franco P, Grauvogel J. Malignant transformation of a dysembryoplastic neuroepithelial tumor (DNET) characterized by genome-wide methylation analysis. J Neuropathol Exp Neurol. 2016. 75: 358-65
8. Hirose T, Scheithauer BW, Lopes MB, Gerber HA, Altermatt HJ, VandenBerg SR. Ganglioglioma: An ultrastructural and immunohistochemical study. Cancer. 1997. 79: 989-1003
9. Isler C, Cetin OE, Ugurlar D, Ozkara C, Comunoglu N, Kizilkilic O. Dysembryoplastic neuroepithelial tumours: Clinical, radiological, pathological features and outcome. Br J Neurosurg. 2018. 32: 436-41
10. Luyken C, Blumcke I, Fimmers R, Urbach H, Wiestler OD, Schramm J. Supratentorial gangliogliomas: Histopathologic grading and tumor recurrence in 184 patients with a median follow-up of 8 years. Cancer. 2004. 101: 146-55
11. Luzzi S, Elia A, Del Maestro M, Elbabaa SK, Carnevale S, Guerrini F. Dysembryoplastic neuroepithelial tumors: What you need to know. World Neurosurg. 2019. 127: 255-65
12. Nguyen HS, Doan N, Gelsomino M, Shabani S. Dysembryoplastic neuroectodermal tumor: An analysis from the surveillance, epidemiology, and end results program, 2004-2013. World Neurosurg. 2017. 103: 380-5
13. Perkins OC. Ganglioglioma. Arch Pathol Lab Med. 1926. 2: 11
14. Prayson RA, Khajavi K, Comair YG. Cortical architectural abnormalities and MIB1 immunoreactivity in gangliogliomas: A study of 60 patients with intracranial tumors. J Neuropathol Exp Neurol. 1995. 54: 513-20
15. Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol. 2016. 131: 833-45
16. Rivera B, Gayden T, Carrot-Zhang J, Nadaf J, Boshari T, Faury D. Germline and somatic FGFR1 abnormalities in dysembryoplastic neuroepithelial tumors. Acta Neuropathol. 2016. 131: 847-63
17. Slegers RJ, Blumcke I. Low-grade developmental and epilepsy associated brain tumors: A critical update 2020. Acta Neuropathol Commun. 2020. 8: 27
18. Stone TJ, Rowell R, Jayasekera BA, Cunningham MO, Jacques TS. Review: Molecular characteristics of long-term epilepsy-associated tumours (LEATs) and mechanisms for tumour-related epilepsy (TRE). Neuropathol Appl Neurobiol. 2018. 44: 56-69
19. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005. 352: 987-96
20. Thom M, Blumcke I, Aronica E. Long-term epilepsy-associated tumors. Brain Pathol. 2012. 22: 350-79
21. Wolf HK, Muller MB, Spanle M, Zentner J, Schramm J, Wiestler OD. Ganglioglioma: A detailed histopathological and immunohistochemical analysis of 61 cases. Acta Neuropathol. 1994. 88: 166-73
22. Yust-Katz S, Anderson MD, Liu D, Wu J, Yuan Y, Olar A. Clinical and prognostic features of adult patients with gangliogliomas. Neuro Oncol. 2014. 16: 409-13

