

- Department of Neurosurgery, Keio University School of Medicine, Tokyo 160-8582, Japan
Correspondence Address:
Shunsuke Shibao
Department of Neurosurgery, Keio University School of Medicine, Tokyo 160-8582, Japan
DOI:10.4103/2152-7806.170459
Copyright: © 2015 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Shibao S, Toda M, Yoshida K. Giant cell tumors of the clivus: Case report and literature review. Surg Neurol Int 25-Nov-2015;6:
How to cite this URL: Shibao S, Toda M, Yoshida K. Giant cell tumors of the clivus: Case report and literature review. Surg Neurol Int 25-Nov-2015;6:. Available from: http://surgicalneurologyint.com/surgicalint_articles/giant-cell-tumors-of-the-clivus-case-report-and-literature-review/
Abstract
Background:Clival giant cell tumors (GCTs) are extremely rare with only eight cases reported to date, and malignant transformation is quite rare. Herein, we report a case of an uncontrolled clival GCT, which was transformed malignant, and review the literature.
Case Description:A 25-year-old man experienced double vision for 1 month. Computed tomography and magnetic resonance imaging revealed a clival tumor. The endonasal endoscopic transsphenoidal approach (EEA) was used, and partial resection was performed because of massive bleeding. Histological examination showed a GCT. After radiation therapy, the tumor recurred; the EEA and the anterior transpetrosal approaches were used to perform second and third operations, respectively. The MIB-1 index increased from 4.2% to 26.3%.
Conclusions:GCTs are difficult to treat because of their location, vascularity, and the potential for malignant transformation.
Keywords: Clivus, giant cell tumor, malignant transformation
INTRODUCTION
Giant cell tumors (GCTs) are generally benign, locally aggressive lesions that are typically located in the metaphysis of long bones. GCTs of the skull are rare and constitute <1% of all reported bone GCTs. These tumors preferentially involve the sphenoid and temporal bones.[
CASE DESCRIPTION
A 25-year-old man experienced double vision for 1 month. He had no history of trauma or surgery. Physical examination revealed overall good health. Neurological examinations revealed right abducens nerve palsy. Motor and sensory examinations including cerebellar tests were normal with full cooperation and orientation. Results from laboratory tests conducted on admission, which included blood biochemical analysis, complete blood count, pituitary function, and tumor markers, were normal.
Computed tomography (CT) demonstrated a homogeneously enhanced mass (5.1 cm × 3.1 cm × 4.9 cm) in the clivus [Figure
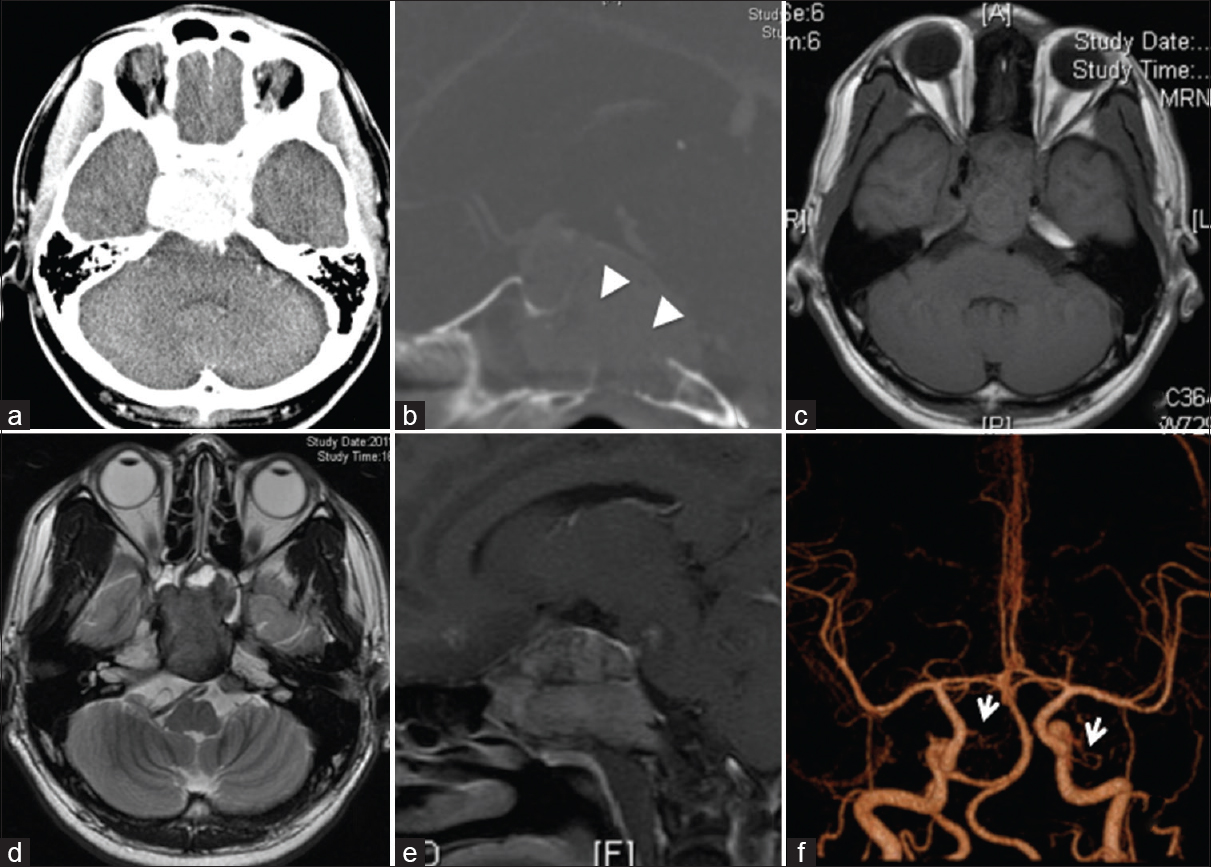
Figure 1
(a) Postcontrast head computed tomography scan revealing an enhanced mass in the clivus. (b) Postcontrast sagittal computed tomography showing clival erosion (arrowheads). T1-weighted (c) and T2-weighted (d) magnetic resonance images showing an iso- and hypo-intense mass, respectively. (e) Postcontrast sagittal T1-weighted magnetic resonance images revealing a homogeneously enhanced tumor. (f) Three-dimensional angiography showing both meningohypophyseal trunks (arrows)
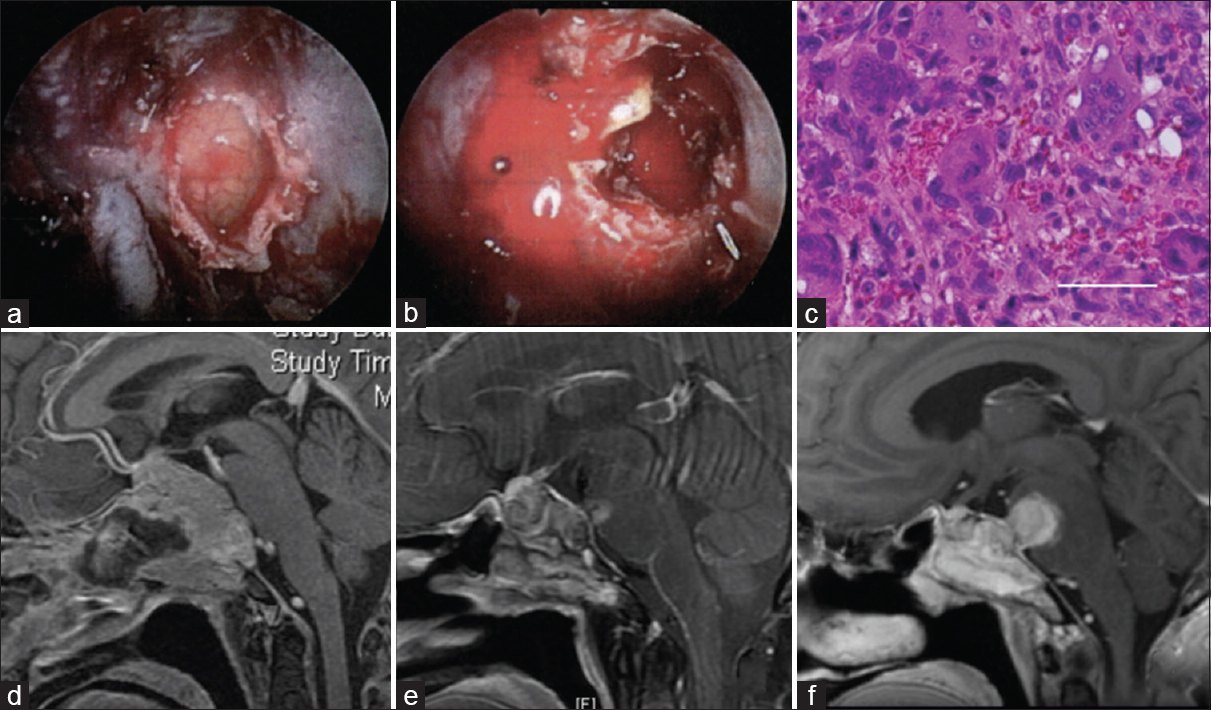
Figure 2
The first operation via an endoscopic endonasal transsphenoidal approach. An intraoperative image showing a yellowish gray tumor of the clivus (a), and profuse bleeding in the tumor (b). (c) A histological specimen showing numerous multinucleated giant cells with dense eosinophilic cytoplasm, scattered against a background of uniform mononuclear cells. Bar 50 μm. (d) Postcontrast sagittal T1-weighted magnetic resonance images revealing a partially resected tumor via the endoscopic endonasal transsphenoidal approach. Regrowth of the tumor after radiation (e) and showing an increase in recurrent tumor size (f)
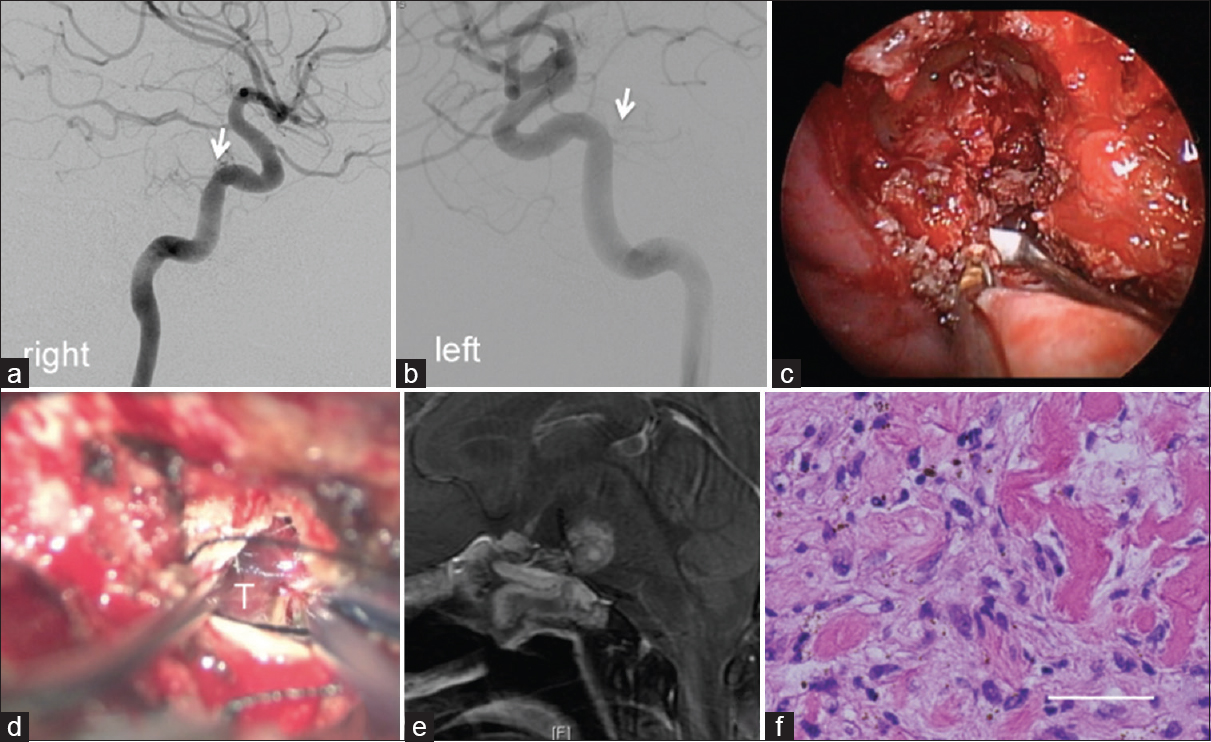
Figure 3
Angiography showing weak tumor staining from meningohypophyseal trunks (arrows). Right side (a), and left side is (b). The second (c) and the third operations (d): An intraoperative image showing a fibrous tumor via the endoscopic endonasal transsphenoidal approach (c), and the anterior transpetrosal approach (d), respectively. (e) Postcontrast sagittal T1-weighted magnetic resonance images revealing partial resection after surgery. (f) A histological specimen showing spindle cell proliferation, with scattered giant cells, pigmentation, and hyalinization. Bar 50 μm
Informed consent was obtained from this patient.
DISCUSSION
Only 3–7% of all primary bone neoplasms manifest as GCTs, which are relatively rare.[
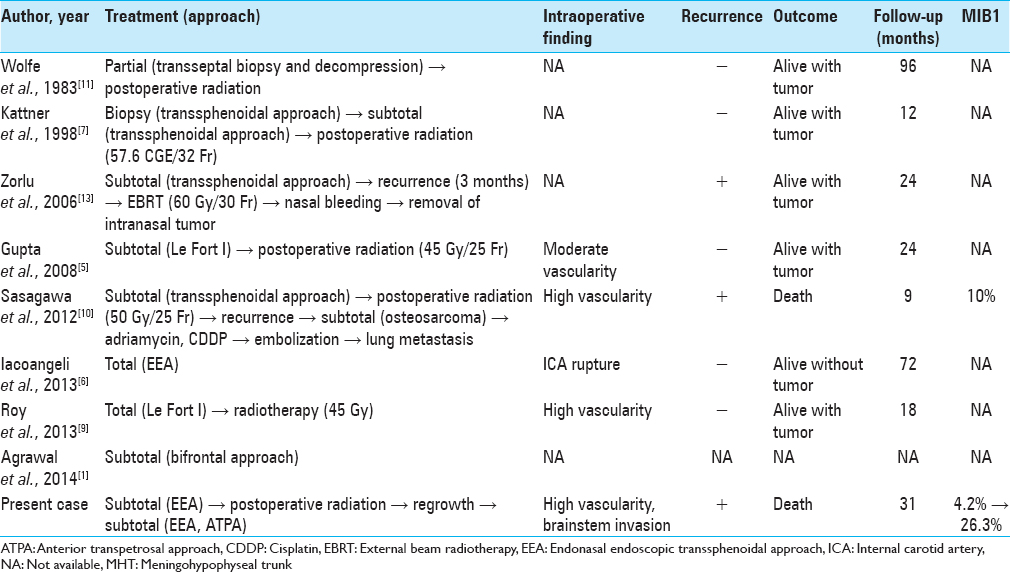
Some papers reported that total resection could enable a reduction in GCT recurrence.[
Radiation therapy has been performed for seven residual tumors. In most cases, the tumor was stable for >1 year. However, two cases including ours showed malignant transformation, leading to resistance to radiation therapy. Sasagawa et al. reported a secondary malignant clival GCT with an MIB-1 index of 10%.[
Chemotherapy has not been performed for clival GCTs and has rarely been evaluated in other skull base GCTs. Yamamoto et al. reported two cases of cranial base GCTs that responded well to chemotherapy and showed regression on periodic CT scans.[
CONCLUSIONS
It is challenging to treat clival tumors because of their location, vascularity, and potential malignant transformation. Angiographic assessment is important in order to control intraoperative bleeding. Pathological assessments such as the MIB-1 index are required for further investigation of the biological properties of clival GCTs.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Agrawal A, Gali R, Shanthi V, Ramakrishna BA, Mohan KV. Giant cell tumor of the clivus with presence of epithelioid histiocytes. Asian J Neurosurg. 2014. 9: 48-9
2. Chawla S, Henshaw R, Seeger L, Choy E, Blay JY, Ferrari S. Safety and efficacy of denosumab for adults and skeletally mature adolescents with giant cell tumour of bone: Interim analysis of an open-label, parallel-group, phase 2 study. Lancet Oncol. 2013. 14: 901-8
3. Epstein N, Whelan M, Reed D, Aleksic S. Giant cell tumor of the skull: A report of two cases. Neurosurgery. 1982. 11: 263-7
4. Goldenberg RR, Campbell CJ, Bonfiglio M. Giant-cell tumor of bone. An analysis of two hundred and eighteen cases. J Bone Joint Surg Am. 1970. 52: 619-64
5. Gupta R, Mohindra S, Mahore A, Mathuriya SN, Radotra BD. Giant cell tumour of the clivus. Br J Neurosurg. 2008. 22: 447-9
6. Iacoangeli M, Di Rienzo A, Re M, Alvaro L, Nocchi N, Gladi M. Endoscopic endonasal approach for the treatment of a large clival giant cell tumor complicated by an intraoperative internal carotid artery rupture. Cancer Manag Res. 2013. 5: 21-4
7. Kattner KA, Stroink A, Gupta K, Fukushima T, Li C. Giant cell tumor of the sphenoid bone. Skull Base Surg. 1998. 8: 93-7
8. Martins AN, Dean DF. Giant cell tumor of sphenoid bone: Malignant transformation following radiotherapy. Surg Neurol. 1974. 2: 105-7
9. Roy S, Joshi NP, Sigamani E, Malik A, Sharma MC, Mohanti BK. Clival giant cell tumor presenting with isolated trigeminal nerve involvement. Eur Arch Otorhinolaryngol. 2013. 270: 1167-71
10. Sasagawa Y, Tachibana O, Shiraga S, Takata H, Kinoshita E, Nojima T. Secondary malignant giant cell tumor of the clivus: Case report. Clin Neurol Neurosurg. 2012. 114: 786-8
11. Wolfe JT, Scheithauer BW, Dahlin DC. Giant-cell tumor of the sphenoid bone. Review of 10 cases. J Neurosurg. 1983. 59: 322-7
12. Yamamoto M, Fukushima T, Sakamoto S, Tomonaga M. Giant cell tumor of the sphenoid bone: Long-term follow-up of two cases after chemotherapy. Surg Neurol. 1998. 49: 547-52
13. Zorlu F, Selek U, Soylemezoglu F, Oge K. Malignant giant cell tumor of the skull base originating from clivus and sphenoid bone. J Neurooncol. 2006. 76: 149-52

