

- Department of Endocrinology, Post Graduate Institute of Medical Education and Research, Chandigarh, India.
- Department of Neurosurgery, Post Graduate Institute of Medical Education and Research, Chandigarh, India.
- Department of Histopathology, Post Graduate Institute of Medical Education and Research, Chandigarh, India.
- Department of Nuclear Medicine, Post Graduate Institute of Medical Education and Research, Chandigarh, India.
Correspondence Address:
Pinaki Dutta, Professor, Department of Endocrinology, Post Graduate Institute of Medical Education and Research, Chandigarh, India
DOI:10.25259/SNI_1013_2022
Copyright: © 2022 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Ananda Mohan Chakraborty1, Sushant Kumar Sahoo2, Debajyoti Chatterjee3, Pinaki Dutta1, Rajender Kumar4, Sanjay Kumar Bhadada1. IgG4-related hypophysitis: A monocentric experience from North India. 16-Dec-2022;13:578
How to cite this URL: Ananda Mohan Chakraborty1, Sushant Kumar Sahoo2, Debajyoti Chatterjee3, Pinaki Dutta1, Rajender Kumar4, Sanjay Kumar Bhadada1. IgG4-related hypophysitis: A monocentric experience from North India. 16-Dec-2022;13:578. Available from: https://surgicalneurologyint.com/surgicalint-articles/12063/
Date of Submission
04-Nov-2022
Date of Acceptance
29-Nov-2022
Date of Web Publication
16-Dec-2022
Abstract
Background: Immunoglobulin (Ig)G4-related disease is a systemic fibroinflammatory disease characterized by dense infiltration of IgG4-positive plasma cells in the affected tissue(s) with or without elevated plasma levels of IgG4. Hypophysitis itself is a very rare disease with reported prevalence in the operative specimens are around 0.2–0.88%. IgG4-related hypophysitis (IgG4-RH) may account for a substantial percentage of cases previously regarded as idiopathic hypophysitis.
Methods: This study is a registry-based, retrospective, and cohort study from a tertiary care hospital in North India. The medical records and clinical data of biopsy-proven and suspected IgG4-RH patients registered were retrospectively analyzed. Treatment outcome of cases was also explored during this analysis.
Results: Two thousand and six sellar area space-occupying lesions have been operated-on since 2006, among them only four patients had IgG4-RH on histopathological specimen. One case was diagnosed on clinical suspicion. Mean age of the patients was 31.8 ± 6.32 years. Most frequent presenting complaint was headaches. Extracranial manifestations were present in four patients. The most common pituitary dysfunction was cortisol deficiency. 18 F-fluorodeoxyglucose positron emission tomography (18F FDG PET) was helpful in three cases for diagnosis of hypophysitis and other organ involvement. Classical histological findings with storiform fibrosis, obliterative phlebitis seen in two cases, and IgG4-positive plasma cell infiltration were positive in four cases. Surgery was the primary modality of treatment in all four cases. Only one patient received steroids as a primary therapeutic modality.
Conclusion: IgG4-RH is rare. High index of suspicion is required to diagnosis the case precisely. FDG PET is helpful in diagnosing hypophysitis and extrapituitary lesions.
Keywords: Autoimmune hypophysitis, Immunoglobulin G4-related disease, Immunoglobulin G4-related hypophysitis, Pituitary stalk thickening
INTRODUCTION
Immunoglobulin G 4-related disease (IgG4-RD) is a systemic fibroinflammatory disease characterized by dense infiltration of immunoglobulin (Ig)G4-positive plasma cells in the affected tissue(s) with or without elevated plasma levels of IgG4.[
The incidence of IgG4-RD remains unknown. This is due to challenges in making the diagnosis as the disease is often unrecognized or misdiagnosed due to its rarity. The disease mostly occurs in an elderly male patient with a range of 5th–7th decade. Male to female ratio ranges from 1.6: 1 to 4:1 in different cohorts.[
Autoimmune hypophysitis itself is a very rare disease, reported prevalence in the operative specimens is around 0.2–0.88%.[
Objective
The aim of the study was to describe the clinical manifestation, management, and outcome of IgG4 related hypophysitis (IgG4-RH), retrospectively.
Design and setting
A registry-based, retrospective, and cohort study from a tertiary care hospital in North India.
MATERIALS AND METHODS
The medical records and clinical data of biopsy-proven and suspected IgG4-RH patients registered in the Postgraduate Institute of Medical Education and Research, Chandigarh between 2006 and 2022 were retrospectively analyzed. Treatment outcome of cases was also explored during this retrospective analysis. Each and every case was dealt with by a multidisciplinary team, consisting of an endocrinologist, a neurosurgeon, a pathologist, and a radiologist. Demographic data, clinical presentation, radiological data, hormonal profile, and serum IgG4 level were assessed at the presentation. Posttherapy visual and endocrinological outcomes and imaging features were analyzed. A trend of demography, presentation, biochemical, and imaging characteristics has been discussed as a cluster. Further brief description of each case has been made along with treatment and outcome till the last follow-up. Basic statistical methods were applied only as the cumulative number of cases was too small for any advance statistics.
RESULTS
Two thousand and six sellar area space-occupying lesions (SOL) have been operated-on since 2006, among them only four patients had histopathology proven IgG4-RH. One case was diagnosed on clinical suspicion, which was presented with pituitary stalk thickening and was not biopsy-proven. After considering all organ manifestations, 154 biopsy-proven IgG4-RD had been reported from this institute.
Three of those five patients were female. The mean age of the patients was 31.8 ± 6.32 years, varied from 16 years to 38 years in this cohort. The median duration of symptoms was 12 months (6 months–20 years).
Most frequent presenting complaint was headaches seen in three cases. Among female patients, all three of them had menstrual abnormality in the form of amenorrhea or oligomenorrhea. Infertility after 17 years of marriage was the presenting complaint in one female patient. Other manifestations were short stature, diminution of vision, and poor development of secondary sexual characteristics.
Extracranial manifestations were present in five patients, consisting of pelvic adhesion, pancreatitis, retroperitoneal fibrosis leading to hydronephrosis and renal failure, and primary testicular failure in one patient each. Adamantinomas craniopharyngioma was associated with IgG4-RD in one patient.
The most common pituitary dysfunction was cortisol deficiency, which was presented in three out of those five patients, followed by hyperprolactinemia present in two patients. One patient presented with central diabetes insipidus and one had primary hypogonadism.
Sellar SOL was the imaging findings in three of the patients, one patient presented with only supra sellar SOL, thickening of the pituitary stalk was present in four of the patients, and one of them had only thickened stalk as the sole imaging finding. One of the patients had craniopharyngioma, loss of pituitary bright spot was evident only in one patient.
The maximum tumor volume reported was 21.57 cc, cavernous sinus extension was present in one patient. Classical histological findings with storiform fibrosis, obliterative phlebitis present in two cases, and IgG4-positive plasma cell infiltration were positive in four cases. IHC for IgG4 cells was positive in four cases with two cases having IgG4/IgG index >40%.
Serum IgG4 was elevated in two cases, one of them was not biopsy-proven. That case was diagnosed radiologically with magnetic resonance imaging (MRI) and 18 F-fluorodeoxyglucose positron emission tomography-computed tomography (18F FDG PET-CT) which was suggestive of stalk thickening and elevated serum IgG4 level and treatment response. Surgery was the primary modality of all four cases, followed by anti-inflammatory therapy. Only one patient received steroids as a primary therapeutic modality. All 5-patient had stable disease with assigned therapy till the publication of this data. One patient diagnosed on the basis of clinical suspicion and IgG4 level showed satisfactory outcomes in her pituitary hormone profile and menstruation cycle following therapy. Although the desired outcome in the form of fertility was not achieved.
Individual case reports
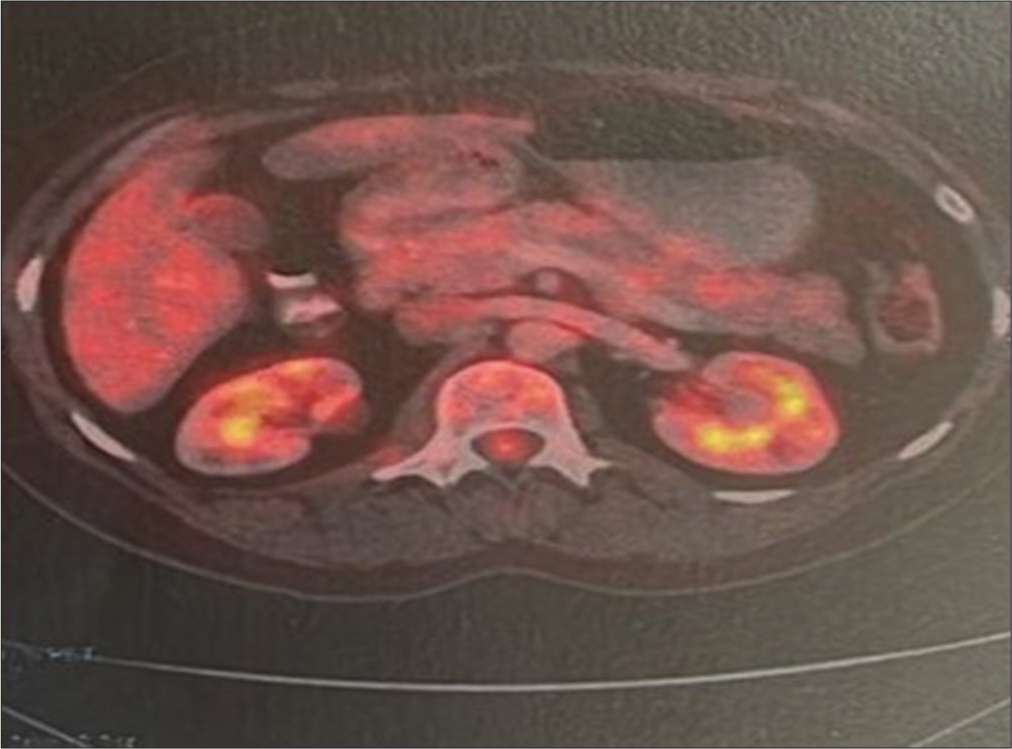
Patient 1, presented with 6-month history of headache and oligomenorrhea. After 4 months of initial complaints, she developed diplopia both in primary gaze and in lateral gaze. Anterior pituitary hormone profile was normal with no features suggestive of diabetes insipidus. Visual field assessment and fundoscopy were normal. Structural imaging suggested altered posterior pituitary bright spot with a sellar SOL measuring 13.5 × 20 mm, with supra-sellar extension with thickening of pituitary stalk. Her excision biopsy specimen was positive for lymphoplasmacytic infiltration, but at that time, IgG4-RD was not suspected due to lack of knowledge of the condition. Nearly 2 years after initial presentation, she developed accelerated hypertension with fatigue and low-grade fever. Laboratory assessment revealed elevated urea and creatinine with ultrasonographic evidence of retroperitoneal fibrosis leading to hydronephrosis with postrenal Acute kidney injury. The diagnosis was subsequently confirmed by 18 F FDG PET-CT. Her pituitary biopsy was reviewed and finally retrospectively diagnosed as IgG4-RD. Later on, she was put on pulse methyl prednisolone followed by maintenance dose of oral prednisolone on which she was responded. (This case has already been published as case report elsewhere).[
Patient 2 presented with headache and amenorrhea for 1 year before contact to this hospital and treated as migraine elsewhere. She had high prolactin level with low cortisol and normal thyroid and gonadotrophin levels. Her visual acuity, fundoscopy, and field of vision were normal. Structural imaging showed a sellar mass of 1.8 × 1.3 × 1.3 cm with suprasellar extension and impingement on right sided optic chiasma, there was evidence of hemorrhage with in the lesion and posterior bright spot was intact [
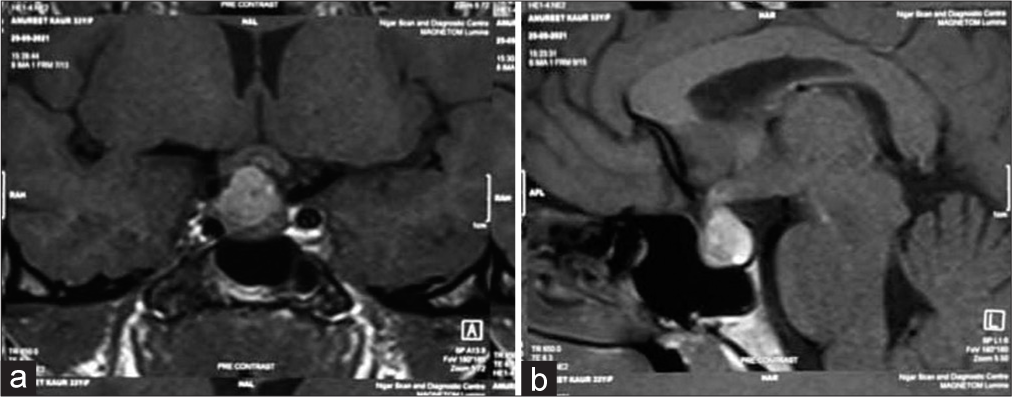
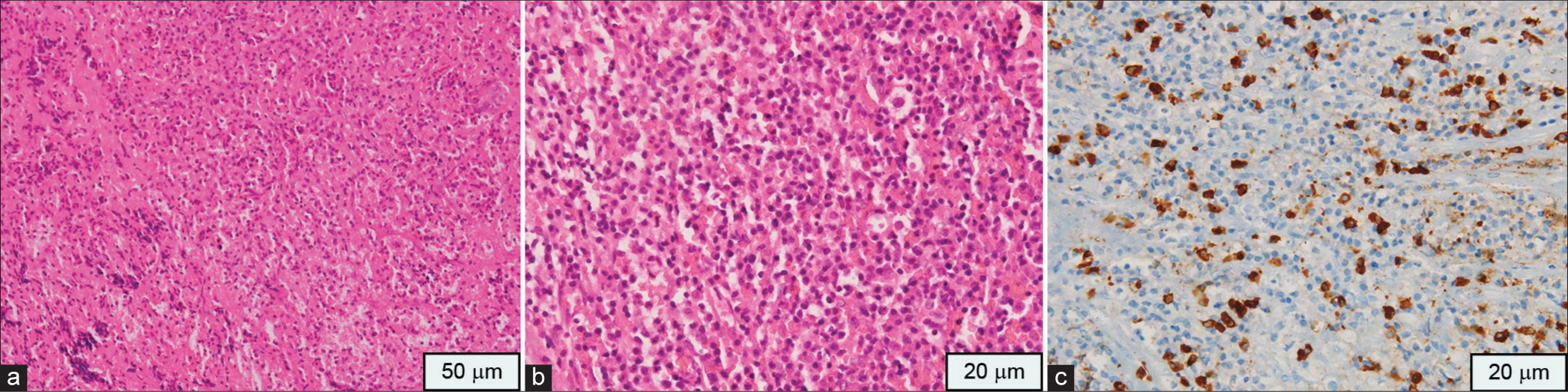
Figure 2:
(a) Biopsy shows pituitary parenchyma with heavy inflammatory infiltrate (Hematoxylin and Eosin, ×100), (b) The inflammation is comprising of numerous plasma cells and histiocytes (Hematoxylin and Eosin, ×200). (c) Many IgG4 positive plasma cells are seen in brown stain (Immunohistochemistry, ×400).
Patient 3 had progressive diminution of vision in the right eye associated with intermittent headache of 1 year duration. He also noticed deceleration in growth velocity, and the lack of development of secondary sexual characteristics. He had panhypopituitarism and visual problem in the form of right sided optic atrophy. MRI of sella showed a 3.8 × 3.5 × 3.1 cm mass with thickening of pituitary stalk and intact posterior bright spot. Excision biopsy showed adamantinomatous craniopharyngioma with part of pituitary and dura showing lymphoplasmacytic infiltration with immunohistochemistry positive for IgG4 plasma cells and fibrosis (This case is published as case report in elsewhere).[
Patient 4 was 34-year-old male. He was non-diabetic and had nasal obstruction with bilateral progressive loss of vision. He was also complaining of short stature and hearing impairment. He had low testosterone with elevated luteinizing hormone and follicle-stimulating hormone and low insulin-like growth factor-1. MRI showed an SOL of 1.8 × 3.3 × 1.7 cm, intensely enhancing, lobulated, suprasellar, and left para-sellar mass involving the pituitary stalk and infundibulum. There was the involvement of cavernous sinus too. Immunohistochemistry of excision biopsy specimen revealed 20–25 IgG4- positive plasma cells/high-power field, with an IgG4 to IgG ratio of >40%. A section from the dura overlying the sphenoid bone also revealed similar findings. IgG4 immunostaining highlighted 15–17 cells/high-power field (21% of total IgG-positive cells). His serum IgG4 level was 225 mg/dl (This case is published as case report in elsewhere).[
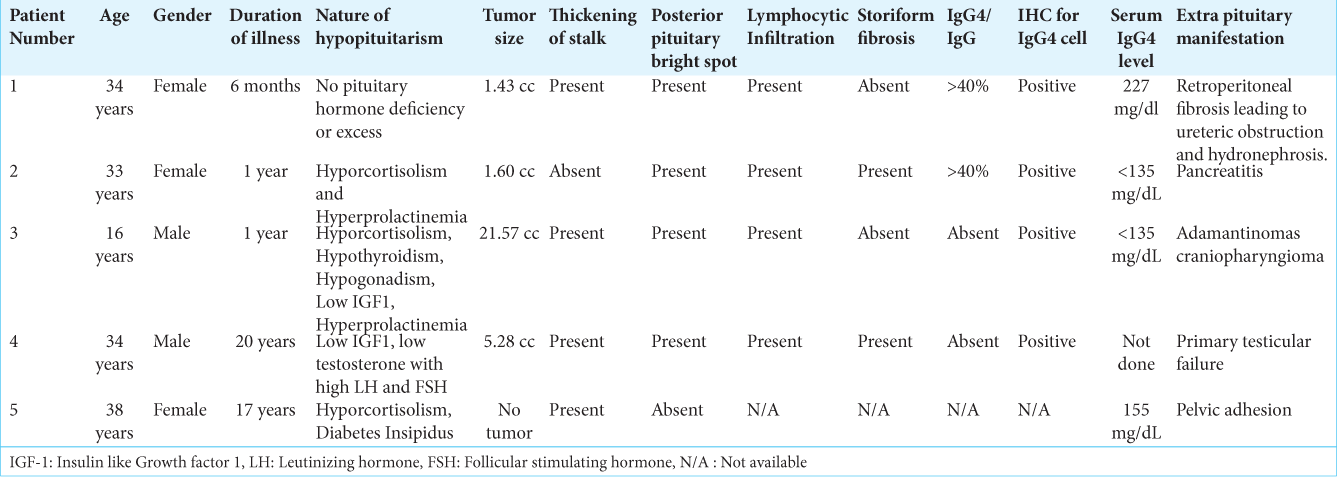
Patient 5 had infertility and oligomenorrhea at presentation. Her symptoms were thought to be contributed by hyperprolactinemia, pelvic adhesion, and ovarian dysfunction. Her MRI and 18 F FDG PET-CT showed thickening of the stalk with normal remaining pituitary. Serum IgG4 level was >135 mg/dL. She was put on Prednisolone 1 mg/kg body weight as an inflammatory dose with gradual tapering. She showed a satisfactory response and resume her menstruation. Summary of patients depicted in
DISCUSSION
The prevalence of IgG4-RH was 0.2% among patient presented with sellar area SOL in our study. There was slight female preponderance as three out of five patients were female.
Mean age of presentation of IgG4-RH was 31.8 ± 6.32 years, which is far younger than the most of the described cohorts. Most common clinical presentation of IgG4-RH was headache in our cohort which was similar to the findings of the previous studies.[
There is no fixed pattern of pituitary deficiency described in the literature in relation to IgG4-RH.[
Our study was in contrary to most of the cohort that showed a median age of presentation of 6th decade with male preponderance with ration ranging from 1.6:1 to 4:1.[
In contrary to our study, Bhargava et al. found different results, where they analyzed a cohort of eight patients, finding as the most common hormone deficiency being thyroid stimulating hormone (TSH), followed by ACTH and then gonadotrophin deficiency. In their cases, postoperative endocrine function did not improve and most of their patients permanently required pituitary hormone replacement therapy.[
Except that fifth case, all other cases were diagnosed retrospectively after neurosurgical intervention and histopathology findings. Only fifth case was suspected clinically and a therapeutic trial of prednisolone was given with satisfactory response. Indication for neurosurgical intervention was similar for all other sellar and supra sellar lesion, such as visual impairment, stalk compression, hypofunctioning of pituitary, and diagnostic uncertainty.
Radiologically, MRI is considered to be the best modality of investigation.[
In this present study, except for one case, all other cases had SOL at the sellar region, maximum tumor volume encountered was 21.57 cc, stalk thickening was present in 4 cases (80%), and the posterior pituitary bright spot was absent in one case, which was presented with central diabetes insipidus.
Criteria devised by Leporati et al. which is now widely accepted to diagnose IgG4-RH, have been used in this study. At present, this is the only pituitary-specific clinical diagnostic criteria.[
Four of our patients had biopsy proven disease and they were fitting to the Criteria 1 and the fifth patient was suspected clinically, she had elevated IgG4 levels, thicken pituitary stalk in MRI, and clinical as well as imaging response to glucocorticoids. We have made a diagnosis for the fifth case on the basis of fulfillment of criteria 2, 4, and 5.
If we consider American college of Rheumatology (ACR)/European league against rheumatism (EULAR) classification criteria published in 2019 for IgG4-RD, four out of five cases fulfilled the criteria with a score of >20, only the fifth case was not fulfilling the criteria.
ACR/EULAR 2019 criteria were not specific for IgG4-RH. Considering the rarity of the disease pituitary involvement was not incorporated in the organ system checklist of this criteria.[
Pathogenesis of the condition was unclear but an autoimmune mechanism or chronic infection has been proposed as a possible cause.[
Although there is no standard treatment protocol for IgG4-RD, steroid is considered as a cornerstone of therapy. We have treated our two patients with steroids, one as primary modality and other as second line after excision biopsy. They were started on anti-inflammatory dose of steroid and subsequently tapered off in 6 months. Patient 2 had pancreatitis as a part of systemic manifestation treated with prednisolone. The latest review emphasizes a pituitary biopsy to diagnose IgG4-RH. In all cases of IgG4-RH, steroid sparing agents such as Mycophenolate mofetil and Rituximab may be tried.
Isolated IgG4-RH was seen in 36% cases by Amirbaigloo et al. and in 65% from the German Pituitary Tumor Registry.[
Considering our experience, IgG4-RH is rare and indistinguishable from other causes of hypophysitis unless biopsy is done. As the understanding of IgG4-RH getting better, people considering it as differential diagnosis of hypophysitis. We emphasize a multi-disciplinary approach for proper diagnosis and management of this rare disorder.
Limitation
The shortfalls of our study are retrospective nature and small sample size. Even if this is a monocentric study, our center has a large referral base.
CONCLUSION
IgG4-RH is an incompletely understood rare disease. It is being increasingly recognized due to greater awareness among physicians, serum IgG4 assay, and repeat staining of histological samples previously labeled as unspecified and add into the diagnosis. It is prudent to get an extrapituitary screening imaging done with FDG PET if hypophysitis is suspected. If the serum IgG4 concentration is >140 mg/dL and if trial of glucocorticoids resolves the presenting symptoms, then IgG4-RH is highly probable. In case, there is a failure of resolution of symptoms with glucocorticoids or if there is any surgical indication or diagnostic uncertainty and serum IgG4 concentrations are not elevated, an early pituitary biopsy should be sought. If there is an extrapituitary organ involvement found on initial screening, then a biopsy of the affected organ should be sought to make a diagnosis. FDG PET should be considered as an important modality of investigation in suspected cases of IgG4-RD.
Declaration of patient consent
Patients’ consent not required as patients’ identities were not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
SUPPLEMENTARY TABLE

References
1. Amirbaigloo A, Esfahanian F, Mouodi M, Rakhshani N, Zeinalizadeh M. IgG4-related hypophysitis. Endocrine. 2021. 73: 270-91
2. Arora K, Rivera M, Ting DT, Deshpande V. The histological diagnosis of IgG4-related disease on small biopsies: Challenges and pitfalls. Histopathology. 2019. 74: 688-98
3. Bhansali A, Kumar A, Dutta P, Bhadada S, Khandelwal N, Radotra B. A woman with pituitary mass presenting as oligoamenorrhea and later with acute renal failure. Endocrinologist. 2008. 18: 13-5
4. Bhargava R, Hussein Z, Dorward NL, Grieve JP, Jaunmuktane Z, Marcus HJ. IgG4-related hypophysitis: A retrospective cohort study. Acta Neurochir (Wien). 2022. 164: 2095-103
5. Brito-Zerón P, Ramos-Casals M, Bosch X, Stone JH. The clinical spectrum of IgG4-related disease. Autoimmun Rev. 2014. 13: 1203-10
6. Caranci F, Leone G, Ponsiglione A, Muto M, Tortora F, Muto M. Imaging findings in hypophysitis: A review. Radiol Med. 2020. 125: 319-28
7. Caturegli P, di Dalmazi G, Lombardi M, Grosso F, Larman HB, Larman T. Hypophysitis secondary to cytotoxic T-Lymphocyte-associated protein 4 blockade. Am J Pathol. 2016. 186: 3225-35
8. Caturegli P, Lupi I, Landek-Salgado M, Kimura H, Rose NR. Pituitary autoimmunity: 30 years later. Autoimmun Rev. 2008. 7: 631-7
9. Caturegli P, Newschaffer C, Olivi A, Pomper MG, Burger PC, Rose NR. Autoimmune hypophysitis. Endocr Rev. 2005. 26: 599-614
10. Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012. 25: 1181-92
11. Flanagan DE, Ibrahim AE, Ellison DW, Armitage M, Gawne-Cain M, Lees PD. Inflammatory hypophysitis-the spectrum of disease. Acta Neurochir (Wien). 2002. 144: 47-56
12. Fleseriu M, Hashim IA, Karavitaki N, Melmed S, Murad MH, Salvatori R. Hormonal replacement in hypopituitarism in adults: An endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2016. 101: 3888-921
13. Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001. 344: 732-8
14. Imber BS, Lee HS, Kunwar S, Blevins LS, Aghi MK. Hypophysitis: A single-center case series. Pituitary. 2015. 18: 630-41
15. Jain N, Dutta P, Gupta AK, Bal A, Ahuja C, Bhansali A. A rare cause of sellar-suprasellar mass: Igg4-related infundibulohypophysitis. AACE Clin Case Rep. 2018. 4: e422-6
16. Karadeniz H, Vaglio A. IgG4-related disease: A contemporary review. Turk J Med Sci. 2020. 50: 1616-31
17. Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P. IgG4-related hypophysitis: A new addition to the hypophysitis spectrum. J Clin Endocrinol Metab. 2011. 96: 1971-80
18. Li Y, Gao H, Li Z, Zhang X, Ding Y, Li F. Clinical characteristics of 76 patients with IgG4-related hypophysitis: A systematic literature review. Int J Endocrinol. 2019. 2019: 1-10
19. Pal R, Chatterjee D, Singla R, Jain N, Bhansali A, Dutta P. Co-occurrence of craniopharyngioma and IgG4-related hypophysitis: An epiphenomenon or a mere coincidence?. World Neurosurg. 2020. 136: 193-7
20. Sah RP, Chari ST. Serologic issues in IgG4-related systemic disease and autoimmune pancreatitis. Curr Opin Rheumatol. 2011. 23: 108-13
21. Stone JH, Khosroshahi A, Deshpande V, Chan JKC, Heathcote JG, Aalberse R. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum. 2012. 64: 3061-7
22. Takahashi H, Yamamoto M, Suzuki C, Naishiro Y, Shinomura Y, Imai K. The birthday of a new syndrome: IgG4-related diseases constitute a clinical entity. Autoimmun Rev. 2010. 9: 591-4
23. Wallace ZS, Deshpande V, Mattoo H, Mahajan VS, Kulikova M, Pillai S. IgG4-related disease: Clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheumatol. 2015. 67: 2466-75
24. Wallace ZS, Naden RP, Chari S, Choi H, Della-Torre E, Dicaire J. The 2019 American college of rheumatology/ european league against rheumatism classification criteria for IgG4-related disease. Arthritis Rheumatol. 2020. 72: 7-19
25. Wallace ZS, Zhang Y, Perugino CA, Naden R, Choi HK, Stone JH. Clinical phenotypes of IgG4-related disease: An analysis of two international cross-sectional cohorts. Ann Rheum Dis. 2019. 78: 406-12
26. Warmbier J, Lüdecke DK, Flitsch J, Buchfelder M, Fahlbusch R, Knappe UJ. Typing of inflammatory lesions of the pituitary. Pituitary. 2022. 25: 131-42
27. Yuen KCJ, Moloney KJ, Mercado JU, Rostad S, McCullough BJ, Litvack ZN. A case series of atypical features of patients with biopsy-proven isolated IgG4-related hypophysitis and normal serum IgG4 levels. Pituitary. 2018. 21: 238-46
28. Zen Y, Nakanuma Y. IgG4-related disease. Am J Surg Pathol. 2010. 34: 1812-9

