

- Department of General Surgery, Loma Linda University, California, United States.
- Department of Neuropathology and Human Anatomy, Loma Linda University, California, United States.
- Department of Neurosurgery, Loma Linda University School of Medicine, Loma Linda, California, United States.
Correspondence Address:
Miguel Angel Lopez-Gonzalez, Department of Neurosurgery, Loma Linda University School of Medicine, Loma Linda, California, United States.
DOI:10.25259/SNI_859_2021
Copyright: © 2021 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Julie Mayeku1, Jeremy Deisch2, Miguel Angel Lopez-Gonzalez3. Immunoglobulin G4-related disease of the cavernous sinus with orbit invasion – A case report. 08-Nov-2021;12:557
How to cite this URL: Julie Mayeku1, Jeremy Deisch2, Miguel Angel Lopez-Gonzalez3. Immunoglobulin G4-related disease of the cavernous sinus with orbit invasion – A case report. 08-Nov-2021;12:557. Available from: https://surgicalneurologyint.com/surgicalint-articles/11217/
Date of Submission
25-Aug-2021
Date of Acceptance
19-Oct-2021
Date of Web Publication
08-Nov-2021
Abstract
Background: Immunoglobulin G4-related disease (IgG4-RD) is a rare systemic disease of unknown etiology. It is characterized by tissue infiltration caused by IgG4 plasma cells and sclerosing inflammation of various body organs. At present, there are very few reported cases of IgG4-RD invasion of cavernous sinus and the orbit.
Case Description: A 56-year-old female with a history of rheumatoid arthritis was presented with a gradual onset of right oculomotor, abducens, and trigeminal nerve deficits. Four weeks after the onset of symptoms, the patient developed gradual visual deficit. Following this, a trial of steroids was administered to the patient. However, the treatment did not work as expected and patient’s condition worsened. She progressed on to suffer complete visual loss in the right eye. Extensive work-up conducted on her turned out to be nondiagnostic. After this, the patient was referred to us for our evaluation. Neuroimaging revealed a right-sided cavernous sinus and orbital apex lesion. Given the lack of diagnosis and response to steroid treatment, we recommended surgical intervention and performed a modified pterional and pretemporal approach with extradural anterior clinoidectomy and transcavernous approach. We performed a lesion biopsy and cavernous sinus decompression, which helped in the partial recovery of visual function. The pathology report was consistent with IgG4-RD.
Conclusion: IgG4-RD is a rare disease that occurs even less in combination with cavernous sinus and orbit invasion. The rarity of the disease and the diverse presentation of symptoms have sometimes caused delayed diagnosis and intervention. Patients who failed to respond to conservative management and patients in the fibrotic stage of the disease without other organ involvement may benefit from surgical intervention if amenable. Early suspicion, diagnosis, and intervention can facilitate better prognosis.
Keywords: IgG4-related diseases, IgG4-related disease with intracranial invasion, Immunoglobulin G4-related disease of cavernous sinus and/or orbit invasion
INTRODUCTION
Immunoglobulin G4-related disease (IgG4-RD) is a rare systemic inflammatory disease that is characterized by tissue infiltration caused by IgG4 plasma cells and sclerosing inflammation. The etiology of IgG4-RD is still largely unknown. IgG4-RD was initially perceived to be related to autoimmune pancreatitis. However, recent cases indicate that it can invade anybody organ, including a few cases that have reported cavernous sinus and orbit invasion. The rarity of this disease and more so the rarity of IgG4-RD with cavernous invasion have limited proper characterization of the disease. This is the main reason for the delay in diagnosis and proper management of the disease.[
CASE PRESENTATION
Clinical presentation
A 56-year-old female with a history of rheumatoid arthritis was managed chronically with immunosuppression with etanercept. She developed a preoperative progression of visual decline and ophthalmoplegia for 7 weeks. Initially, she was admitted to the emergency department with right-sided eye pain and diplopia of 2 weeks duration and was hospitalized and managed by neurology, rheumatology, oncology, and ophthalmology. Her vision was initially intact with visual acuity of 20/25 on the right eye and 20/20 on the left eye with complete visual fields. She presented with the right pupil sparing ophthalmoplegia, also with trochlear, and abducens palsy. Specifically, she had right-sided ptosis, limited adduction/abduction, limited down/up gaze, having also ophthalmic (V1) and maxillary (V2) trigeminal nerve sensory branches dysfunction with hypoesthesia. Extensive work-up was conducted, including lumbar puncture with cerebrospinal fluid analysis and cytology studies, which returned normal. The meningitis panel and cultures were all negative. Additional tests such as human immunodeficiency virus, angiotensin-converting enzyme, venereal disease research laboratory, antineutrophil cytoplasmic antibody, antinuclear antibodies, serum protein electrophoresis, immunofixation electrophoresis, and lymphoma profile were all negative. Computed tomography of chest, abdomen, and pelvis did not show any signs of malignancy. Neuroimaging showed enhancing soft tissue within the right cavernous sinus extending into the right orbital apex and posterior aspect of the right lateral rectus muscle, with enlargement of the superior ophthalmic vein [
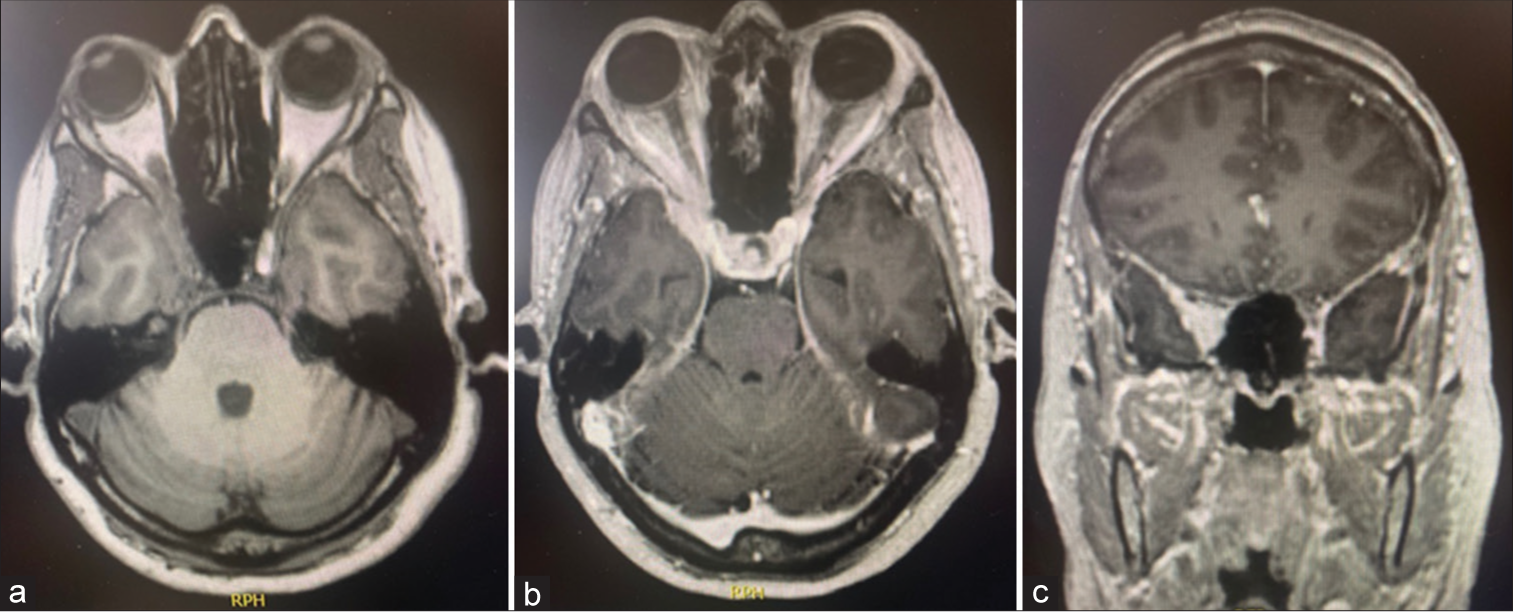
Differential diagnosis suggested retro-orbital granulomatous pseudotumor such as Tolosa-Hunt syndrome, granulomatous infection such as sarcoidosis, infiltrating neoplasm such as lymphoma, metastasis, and other neoplastic etiologies. Cavernous sinus thrombosis and carotid cavernous fistula were considered unlikely, given the lack of conjunctival injection and papilledema.
The patient was initially administered with intravenous steroids for 7 days that improved her ptosis and V1-V2 hypoesthesia, with stable visual function. A repeat brain and orbit MRI 1 week after treatment did not show changes on the right orbital apex/cavernous sinus lesion, but given mild improvement, she was then discharged home with etanercept and methotrexate weekly.
She had outpatient follow-up with ophthalmology 3 weeks after discharge from the hospital showing similar visual examination. One week later, the patient experiences complete right visual loss, then she was referred to neurosurgery clinic and she was evaluated for possible urgent surgical intervention.
The patient underwent to a right-sided skull base pretemporal approach with orbitotomy at the lateral wall and roof of the orbital apex, and extradural anterior clinoidectomy. Mini-rongeurs were used to decompress the optic canal, and a 1 mm diamond drill was used at the core of anterior clinoid process and optic strut. Mini-rongeurs and skull base dissectors were used to remove the cortical bone during the anterior clinoid process. The lateral wall of the cavernous sinus was then explored to allow the dissection of the cranial nerves III, IV, V1, and V2. Samples were taken from the posterior orbital lesion, the orbital apex lesion, and the inflamed tissue around clinoid carotid. The lateral wall of cavernous sinus was opened at the supratrochlear triangle and tissue sample was extracted from there as well. Durotomy was performed for intradural exploration to identify the entire oculomotor nerve through the intradural cistern segment and cavernous sinus and the infiltrated dura in the middle fossa was resected. Fibrin glue, nonsuturable synthetic dura substitute, and fat graft harvested from the abdomen were used for dura reconstruction. Finally, during postoperative period, the patient was administered a short course of steroids.
The resected tissue was submitted to the department of surgical pathology for examination. The dura and soft tissues showed dense fibrosis and a brisk chronic inflammatory infiltrate with numerous lymphocytes, plasma cells, and occasional histiocytes. During immunohistochemistry, numerous plasma cells were highlighted on IgG staining. Approximately half of the plasma cells also expressed IgG4. The combined histomorphology and immunophenotype were consistent with IgG-related sclerosing disease involving orbital region soft tissues and dura mater. No granulomatous inflammation or evidence of a neoplastic process was seen [
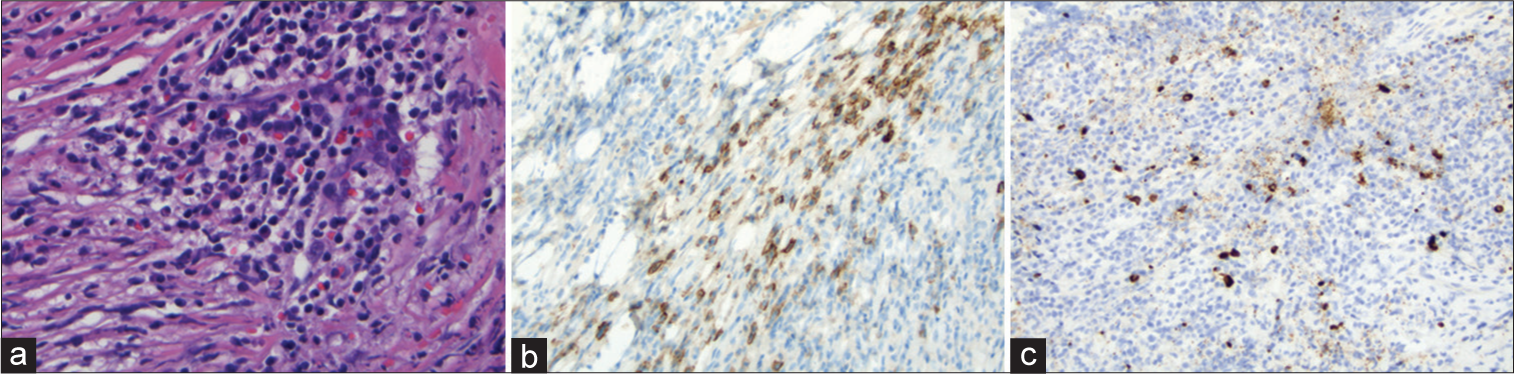
Figure 2:
(a) High-power view showing dense fibrous tissue with lymphocyte and plasma cell-rich inflammatory infiltrate (H&E, ×400); (b) CD138 immunohistochemical stain highlights plasma cells within the inflammatory infiltrate (IHC, ×200); (c) IgG4 immunohistochemical stain stains approximately half of the plasma cells (IHC, ×200).
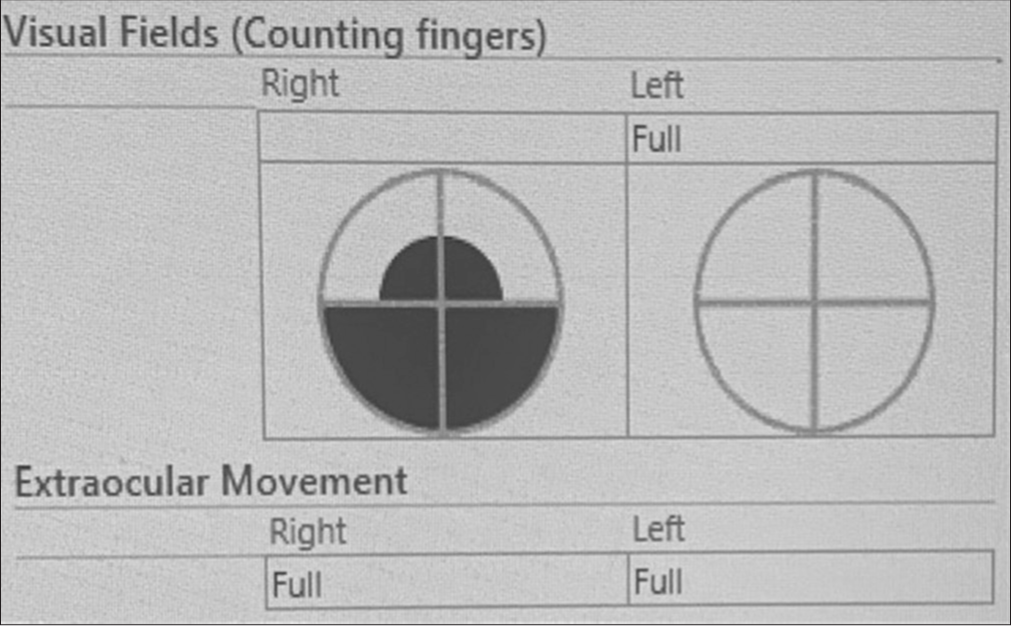
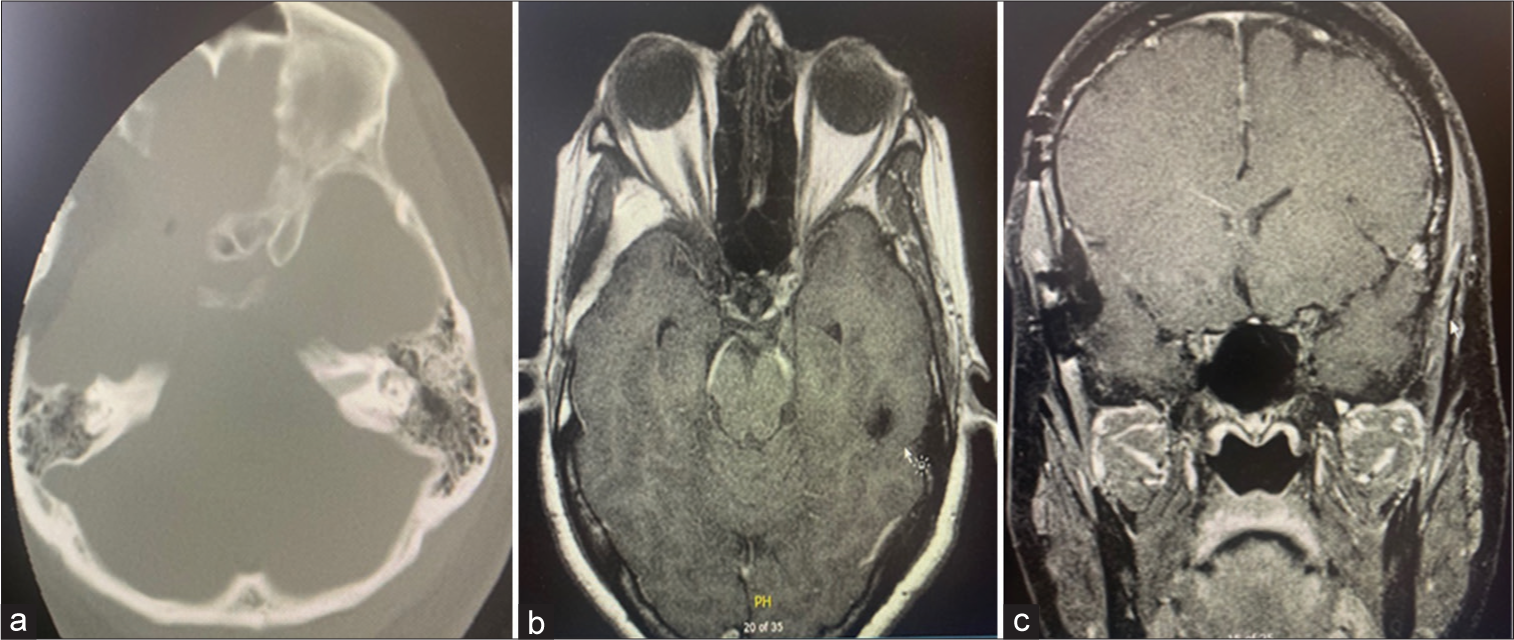
During the 6 months follow-up, extraocular movement deficits from the oculomotor and abducens nerve were resolved. Visual function had also improved; the patient was now able to count fingers from a distance of 3 feet (since she had right complete visual loss preoperatively). Improvement was also noted in the patient’s visual field [
DISCUSSION
IgG4-RD is multiple organs disorder that occurs most frequently in the pancreatic and hepatobiliary system. Isolated head, neck, and brain involvement in IgG4-RD is uncommon. The first case of lateral skull base IgG4-RD was reported in 2012. Isolated IgG4-RD invasion of cavernous sinus is even more rare.[
So far, very few population-based studies of IgG4-related disease have been conducted. Therefore, information about the epidemiology of the disease is only sparsely available. In fact, the current nomenclature of IgG4-RD was accepted as recent as in 2011, while the first international consensus on the pathological findings that currently define IgG4-RD was reached in 2012. The current studies have reported that IgG4-RD is more likely to occur in patients with certain striking demographic features. For instance, IgG4-RD is reported to occur predominantly in men between the sixth and seventh decades of life. It is said to be frequently associated with lymphadenopathy and expected to respond well to steroid therapy. However, these characteristics were not there in the case of our patient. Studies indicate that the pancreas is usually the most affected organ. However, it has also been documented that IgG4-RD can affect nearly every organ in the human body, including the skull base.[
IgG4-RD is diagnosed based on clinical, radiographic, biochemical, and histopathologic evidence. In most cases, IgG4-RD with intracranial invasion occurs with nonspecific symptoms. Neuroimaging findings also mimic other more common intracranial disorders, which can lead to subsequent delays in its diagnosis and management. There are, however, some clinical presentations that are unique to IgG4-RD, such as subacute presentation of clinical symptoms and lack of constitutional illness. Hence, when patients are presented with these conditions, clinicians are advised to include IgG4-RD in the differentials. Fevers and elevations of C-reactive protein levels have also been reported to be unusual in the IgG4-RD patient cohort. This characteristic was consistent with that of our patient who presented a gradual onset of clinical symptoms without constitutional illness, as shown by the unremarkable extensive work-up results.
The most commonly presented symptoms of IgG4-RD are similar to that of any other intracranial space-occupying lesions. The effects of local pressure of the invasion, such as progressive headache, cranial nerve palsies without evidence of systemic disease, nausea, and vomiting, are some of the common symptoms of IgG4-RD. Depending on the location of the disease, isolated lesions often mimicked a diverse spectrum of skull/skull base pathologies including lymphoma, sarcoma, nasopharyngeal carcinoma, neurosarcoidosis, granulomatosis with polyangiitis, sarcoma, giant cell arteritis, Langerhans cell histiocytosis, and benign skull base tumors.[
Radiologically, IgG4-related mass lesions, as in our case, present as homogenously enhanced lesions, which are isointense to hypointense on T1-weighted and T2-weighted images.[
Three histopathological findings characterize the disease in the affected organ: (1) The presence of a storiform pattern of sclerosis; (2) a dense lymphoplasmacytic infiltrate; and (3) an increased proportion of IgG4-positive cells with respect to IgG-positive cells according to immunohistochemical evidence. Although elevated concentrations of IgG4 in tissue and serum are helpful in diagnosing IgG4-RD, neither one is a specific diagnostic marker. In fact, misdiagnoses of IgG4-RD are increasingly common because of the excessive emphasis that physicians tend to place on moderate elevations of serum IgG4 concentration and their overreliance on the finding of IgG4-positive plasma cells in tissues. Histopathological analysis of biopsy specimens remains the cornerstone in the diagnosis of IgG4-RD. Due to the scarcity, ambiguities, and uncommon nature of intracranial IgG4-RD, its diagnosis can be a challenge and may require several biopsies, which can often lead to delays in diagnosis and management of the disease. Corticosteroids and immunosuppressive agents may exacerbate similar appearing lesions such as osteomyelitis or alter the diagnostic yield of a biopsy and therefore should not be started unless there is a suspected diagnosis of IgG4-RD.[
There are currently a few accepted criteria for the diagnosis of IgG4-RD, such as the criteria put forth by Okazaki et al.[
Our patient met the diagnostic criteria of focal lesion in the cavernous sinus and orbit, and had histopathology confirmed through biopsy.
At present, corticosteroids are the first line of treatment administered for IgG4- RD. Proposed regimens include prednisone 40 mg/day with adjustments based on disease response, or prednisolone 0.6 mg/kg/day for 2–4 weeks, followed by a 3–6 months taper to 5 mg/day and maintenance dose of 2.5–5 mg for up to 3 years. Symptomatic improvement, decrease in IgG4 levels, and decrease in lesion load can be expected within weeks of treatment commencement. Biological therapy combined with radiotherapy has also been employed successfully in the induction and maintenance of clinical response to IgG4-RD, and has been proposed as second-line therapy. There is currently no evidence supporting the use of steroid sparing agents to treat this disease.
Successful surgical intervention for treating IgG4-RD has been retrospectively reported in a few studies. However, prospective data on the surgical treatment of IgG4-RD still remain limited. Studies on the natural history of IgG4-RD show that if left untreated, the disease can progress to extensive organ fibrosis. Patients who are in the fibrotic stage of IgG4-RD often respond poorly to steroid therapy, which makes surgical resection a necessary intervention. Our patient exhibited bone and dense fibrous tissue with fibrosis and severe chronic inflammation of IgG4-RD at the orbital apex and cavernous sinus, which severely compressed the optic canal and orbital apex contents. This patient pupil-sparing ophthalmoplegia is an interesting phenomenon. It has been suggested that slow enlarging masses in the cavernous sinus can preserve this function due to the topographic relation and different degrees of compression between parasympathetic pupillomotor fibers, which are not evenly distributed on the surface of the nerve but rather form a distinct bundle in its dorsomedial aspect. Therefore, any compressive force from a mass within the cavernous sinus spares this function and mainly damages the somatosensory axons. Other explanation is based on ischemic changes also due to compression, different to the microvascular ischemic etiology as seen in diabetes mellitus.[
CONCLUSION
IgG4-RD is an uncommon disease that should be included in the differential diagnosis for cavernous sinus lesions. Early suspicion and initiation of therapy can significantly improve the outcome of the treatment for the disease. A delay in treatment for any reason or lack of response can result in serious and irreversible sequelae. As of today, IgG4-RD is treated with systemic corticosteroids at high doses that are subsequently tapered. In cases of IgG4-RD involving cavernous sinus and orbital apex, we strongly recommend that surgical decompression be performed, to prevent permanent deficit that can be caused by inflammatory tissue compression at the compact space between the orbital apex and the optic canal. We believe that more studies should be done in this field, to provide more insight and algorithm on the surgical management of IgG4-RD, especially with skull base involvement.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Ajtai B, Fine E, Lincoff N. Pupil-sparing, painless compression of oculomotor nerve by expanding basilar artery aneurysm. A case of ocular pseudomyasthenia. Arch Neurol. 2004. 61: 1448-50
2. Ardila-Suarez O, Abril A, Gómez-Puerta JA. Enfermedad relacionada con IgG4: Revisión concisa de la literatura. Reumatol Clin. 2017. 13: 160-6
3. Baptista B, Casian A, Gunawardena H, D’Cruz D, Rice CM. Neurological manifestations of IgG4-related disease. Curr Treat Options Neurol. 2017. 19: 14
4. Behbehani RS, Al-Nomas HS, Al-Herz AA, Katchy KC. Bilateral intracranial optic nerve and chiasmal involvement in IgG4-related disease. J Neuroophthalmol. 2015. 35: 229-31
5. Cler SJ, Sharifai N, Baker B, Dowling JL, Pipkorn P, Yaeger L. IgG4-related disease of the skull and skull base-a systematic review and report of two cases. World Neurosurg. 2021. 150: 179-96.e1
6. Goulam-Houssein S, Grenville JL, Mastrocostas K, Munoz DG, Lin A, Bharatha A. IgG4-related intracranial disease. Neuroradiol J. 2019. 32: 29-35
7. Jacobson DM. Relative pupil-sparing third nerve palsy: Etiology and clinical variables predictive of a mass. Neurology. 2001. 56: 797-8
8. Kamisawa T, Okazaki K. Diagnosis and treatment of IgG4-related disease. Curr Top Microbiol Immunol. 2017. 401: 19-33
9. Kanoke A, Ogawa Y, Watanabe M, Kumabe T, Tominaga T. Autoimmune hypophysitis presenting with intracranial multi-organ involvement: Three case reports and review of the literature. BMC Res Notes. 2013. 6: 1-7
10. Karim AF, Bansie RD, Rombach SM, Paridaens D, Verdijk RM, van Hagen PM. The treatment outcomes in IgG4-related disease. Neth J Med. 2018. 76: 275-85
11. Katsura M, Mori H, Kunimatsu A, Sasaki H, Abe O, Machida T. Radiological features of IgG4-related disease in the head, neck, and brain. Neuroradiology. 2012. 54: 873-82
12. Khosroshahi A, Stone JH. A clinical overview of IgG4-related systemic disease. Curr Opin Rheumatol. 2011. 23: 57-66
13. Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol. 2015. 67: 1688-99
14. Kristof RA, Roost DV, Schramm J, Wichers M. Primary granulomatous hypophysitis not responsive to pulsed high dose prednisolone therapy: Case report. J Neurol Neurosurg Psychiatry. 1998. 64: 693-4
15. Kubota T, Moritani S. Orbital IgG4-related disease: Clinical features and diagnosis. ISRN Rheumatol. 2012. 2012: 412896
16. Kuroda N, Inenaga C, Arai Y, Otsuki Y, Tanaka T. Intracranial multiple pseudotumor due to immunoglobulin G4-related disease without other lesions: Case report and literature review. World Neurosurg. 2019. 132: 69-74
17. Lin CK, Lai DM. IgG4-related intracranial hypertrophic pachymeningitis with skull hyperostosis: A case report. BMC Surg. 2013. 13: 1-5
18. Mahajan VS, Mattoo H, Deshpande V, Pillai SS, Stone JH. IgG4-related disease. Annu Rev Pathol. 2014. 9: 315-47
19. Marinelli JP, Marvisi C, Vaglio A, Peters PA, Dowling EM, Palumbo AA. Manifestations of skull base IgG4-related disease: A multi-institutional study. Laryngoscope. 2020. 130: 2574-80
20. McKinney AM, Short J, Lucato L, SantaCruz K, McKinney Z, Kim Y. Inflammatory myofibroblastic tumor of the orbit with associated enhancement of the meninges and multiple cranial nerves. AJNR Am J Neuroradiol. 2006. 27: 2217-20
21. Munawar K, Nayak G, Fatterpekar GM, Sen C, Zagzag D, Zan E. Cavernous sinus lesions. Clin Imaging. 2020. 68: 71-89
22. Nakata R, Yoshimura S, Motomura M, Tsujino A, Hayashi T, Hara M. IgG4-related disease with cavernous sinus and intra-orbital lesions diagnosed by nasal mucosa biopsy. Rinsho Shinkeigaku. 2016. 56: 637-40
23. Okazaki K, Uchida K, Ikeura T, Takaoka M. Current concept and diagnosis of IgG4-related disease in the hepato-biliopancreatic system. J Gastroenterol. 2013. 48: 303-14
24. Peng L, Zhang P, Zhang X, Li J, Zhao J, Liu J. Clinical features of immunoglobulin G4-related disease with central nervous system involvement: An analysis of 15 cases. Clin Exp Rheumatol. 2020. 38: 626-32
25. Ruff MW, Carabenciov ID, Johnson DR, Pollock BE, Parisi JE, Klaas JP. A cavernous sinus lesion clinically responsive to steroids. J Clin Neurosci. 2018. 53: 239-40
26. Singh AD, Soneja M, Memon SS, Vyas S. Interesting case of base of skull mass infiltrating cavernous sinuses. Case Rep. 2016. 2016: bcr2016217669
27. Tanaka T, Fuga M, Teshigawara A, Hasegawa Y, Nishiwaki K, Murayama Y. IgG4-related disease in the frontal convexity concomitant with smoldering multiple myeloma: A case report and review of the literature regarding therapeutic implications. World Neurosurg. 2020. 143: 247-60
28. Tang H, Ding G, Xiong J, Zhu H, Hua L, Xie Q. Clivus inflammatory pseudotumor associated with immunoglobulin G4-related disease. World Neurosurg. 2018. 118: 71-4
29. Tiegs-Heiden CA, Eckel LJ, Hunt CH, Diehn FE, Schwartz KM, Kallmes DF. Immunoglobulin G4-related disease of the orbit: Imaging features in 27 patients. AJNR Am J Neuroradiol. 2014. 35: 1393-7
30. Tomio R, Ohira T, Wenlin D, Yoshida K. Immunoglobulin G4-related intracranial inflammatory pseudotumours along both the oculomotor nerves. BMJ Case Rep. 2013. 2013: bcr2012007320
31. Topiwala K, Hampton C, Boland C, Waitzman D. Neurologic IgG4-related disease. Neurohospitalist. 2019. 9: 118-9
32. Toyoda K, Oba H, Kutomi K, Furui S, Oohara A, Mori H. MR imaging of IgG4-related disease in the head and neck and brain. AJNR Am J Neuroradiol. 2012. 33: 2136-29
33. Umehara H, Okazaki K, Kawa S, Takahashi H, Goto H, Matsui S. The 2020 revised comprehensive diagnostic (RCD) criteria for IgG4-RD. Mod Rheumatol. 2021. 31: 529-33
34. Varrassi M, Gianneramo C, Arrigoni F, Cerrone P, Sucapane P, Marini C. Neurological involvement of IgG4-related disease: Description of a case and review of the literature. Neuroradiol J. 2018. 31: 196-202
35. Wallace ZS, Naden RP, Chari S, Choi HK, Della-Torre E, Dicaire JF. The 2019 American college of rheumatology/European league against rheumatism classification criteria for IgG4-related disease. Arthritis Rheumatol. 2020. 72: 7-19
36. Wick CC, Zachariah J, Manjila S, Brown WC, Malla P, Katirji B. IgG4-related disease causing facial nerve and optic nerve palsies: Case report and literature review. Am J Otolaryngol. 2016. 37: 567-71
37. Yamagishi A, Oshitari T, Tawada A, Baba T, Yamamoto S. The case of IgG4-related ophthalmic disease with perivascular lesions of superior ophthalmic vein associated with optic nerve disturbance. Neuroophthalmology. 2017. 42: 251-5
38. Yeh CH, Tsui YK, Liu H, Chuang SS. Primary IgG4-producing extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) in the cavernous sinus: A mimicker of IgG4-related disease/hypertrophic pachymeningitis. Pathol Int. 2021. 71: 278-80
39. Yoshinaga T, Kurokawa T, Uehara T, Nitta J, Horiuchi T, Sekijima Y. Optic neuropathy from connected intra-and extraorbital lesions in IgG4-related disease. Rinsho Shinkeigaku. 2019. 59: 746-51
40. Zhao Y, Xu J. Imaging features, clinicopathological analysis and diagnostic strategy of IgG4-related hypertrophic pachymeningitis. Ann Palliat Med. 2020. 9: 2551-8

