

- Department of Neurosurgery Fattouma Bourguiba University Hospital, University of Monastir, Monastir, Tunisia.
- Department of Histopathology, Fattouma Bourguiba University Hospital, University of Monastir, Monastir, Tunisia.
Correspondence Address:
Mohamed Amine Hadj Taieb, Department of Neurosurgery, Fattouma Bourguiba University Hospital, University of Monastir, Monastir, Tunisia.
DOI:10.25259/SNI_149_2022
Copyright: © 2022 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Kais Maamri1, Mohamed Amine Hadj Taieb1, Ghassen Elkahla1, Rym Hadhri2, Mehdi Dermoul1. Immunoglobulin G4-related disease presenting as an intracranial mass extended from the infratemporal fossa. 27-May-2022;13:224
How to cite this URL: Kais Maamri1, Mohamed Amine Hadj Taieb1, Ghassen Elkahla1, Rym Hadhri2, Mehdi Dermoul1. Immunoglobulin G4-related disease presenting as an intracranial mass extended from the infratemporal fossa. 27-May-2022;13:224. Available from: https://surgicalneurologyint.com/surgicalint-articles/11619/
Date of Submission
05-Feb-2022
Date of Acceptance
04-May-2022
Date of Web Publication
27-May-2022
Abstract
Background : Neurological manifestations in immunoglobulin G4-related diseases (IgG4-RD) are rare and documented in
Case Description: We present our experience with a biopsy-proven case of IgG4-RD presenting with an intracranial extradural tumor-like mass infiltrating the temporal lobe. The patient was treated with high doses of corticosteroids followed by slow tapering. The neurological manifestations gradually improved and resolved after 2 months with a cerebral MRI showing a significant reduction in the tumoral size.
Conclusion: When it comes to intracranial mass, IgG4-RD neuropathy is one of the rarest differential diagnoses for the central nervous system tumors. Early recognition of IgG4-RD and appropriate establishment of its long-term treatment may avoid unnecessary investigations and morbidity.
Keywords: Corticosteroids, Extra-axial neoplasm, Immunoglobulin G4-related disease, Neurological manifestations, Pseudo-tumors
INTRODUCTION
IgG4-related disease (IgG4-RD) is a recently recognized chronic inflammatory condition and characterized by tissue infiltration with lymphocytes and IgG4-secreting plasma cells, which can involve any organ or system.[
It is a relapsing-remitting disease associated with a tendency to form tissue-destructive lesions. It can damage several sites especially the pancreas, bile ducts, lacrimal, and salivary glands. Head and brain involvement is rare.[
In this article, we present one of the rarest cases of an intracranial tumor-like mass in IgG4-RD.
The purpose of this report is to supply further information helpful in distinguishing this disease from other extra-axial tumors.
CASE PRESENTATION
We report the case of a 37-year-old man with a 6-month history of headaches and blurred vision. Our patient had been followed by an otorhinolaryngologist for 2 years for cervical lymphadenopathy and a right submandibular swelling. The cervical lymphadenopathy biopsy was non-diagnostic twice, showing a non-specific inflammatory disease. He had no other medical background and no personal or familiar history of an autoimmune disease.
On examination, he had significant swelling of the right hemi face and the neck with trismus and a decrease in the visual acuity of the right eye. The dilated fundus examination showed a right papillary paleness.
Peripheral blood markers of inflammation were elevated. Screening for immunodeficiency and mycobacterial infections was negative.
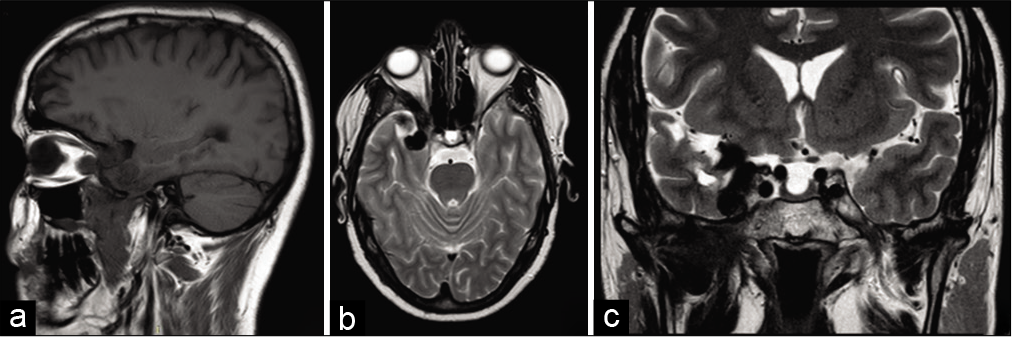
Cerebral MRI showed a pseudotumoral lesion developing in the right pterygoid-palatine fossa spreading to the orbital and the intracranial cavity through the superior orbital fissure. The intracranial portion forms a temporal extra-axial mass mimicking a meningioma that infiltrates the lateral wall of the cavernous sinus. The lesion was strongly enhanced after the injection of gadolinium [
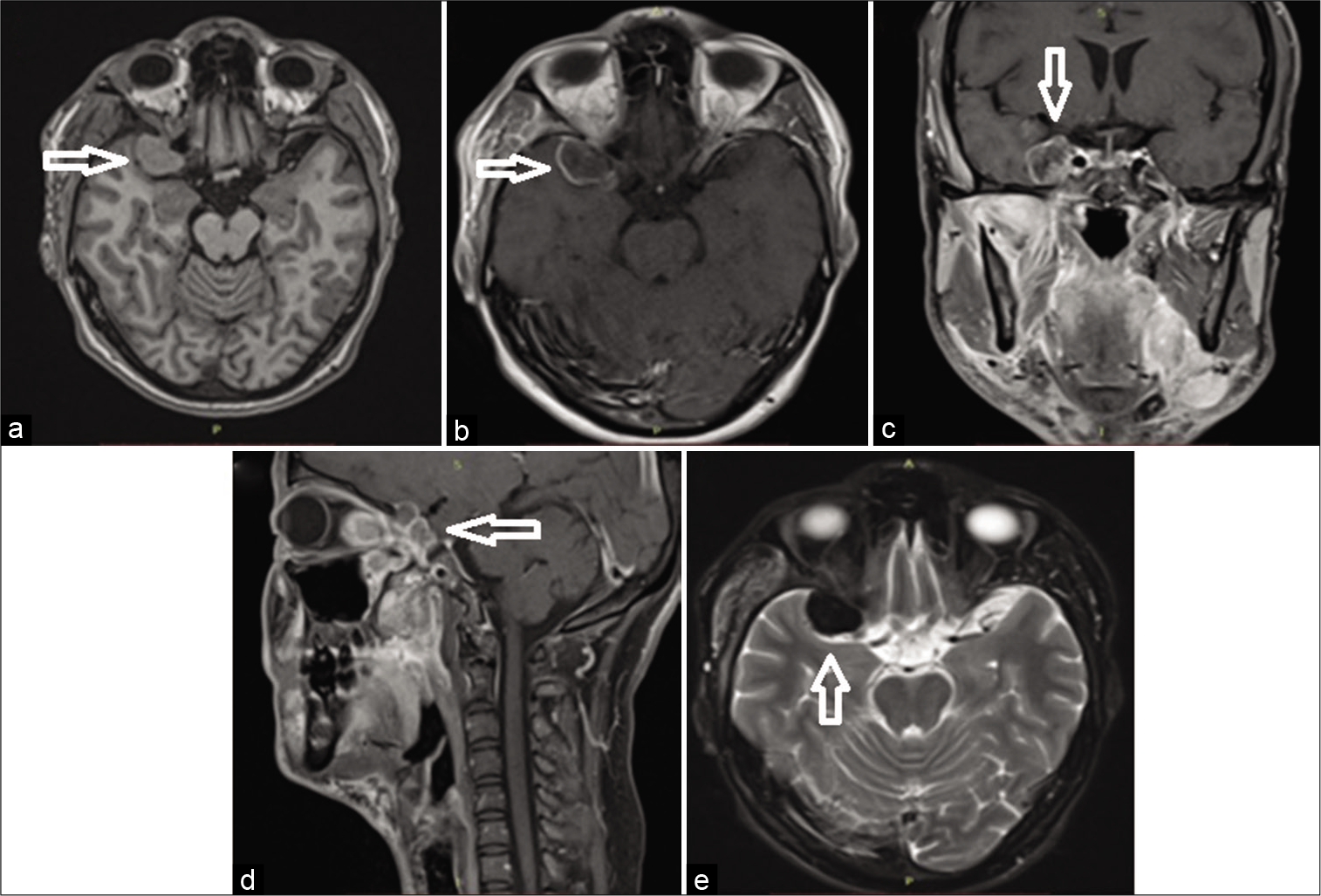
Figure 1:
Preoperative MRI demonstrating a sharply margined extra-axial temporal mass with relatively homogeneous contrast-enhancement and extends to the sella turcica and the sub-temporal fossa (a) T1-weighted image (arrow), (b-d) T1-weighted image with gadolinium contrast on transverse, coronal, and sagittal section, and (e) T2-weighted image (arrow).
The patient was operated through a pterional approach. Our first strategy was a gross total resection of the intracranial portion of the tumor. Regarding its very firm consistency, we opted for a large biopsy of the extra-axial lesion. The tumor was solid, well-delineated, and strongly adherent to the temporal lobe.
Histological examination showed dense lymphoidplasmacytic infiltrate with storiform fibrosis [
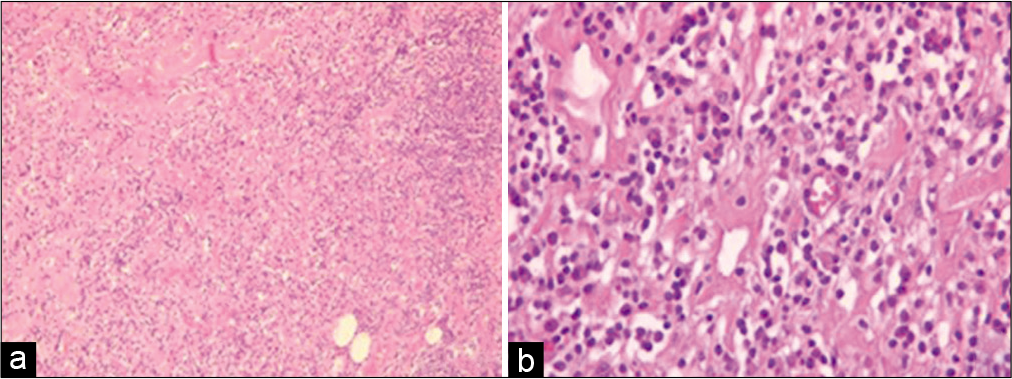
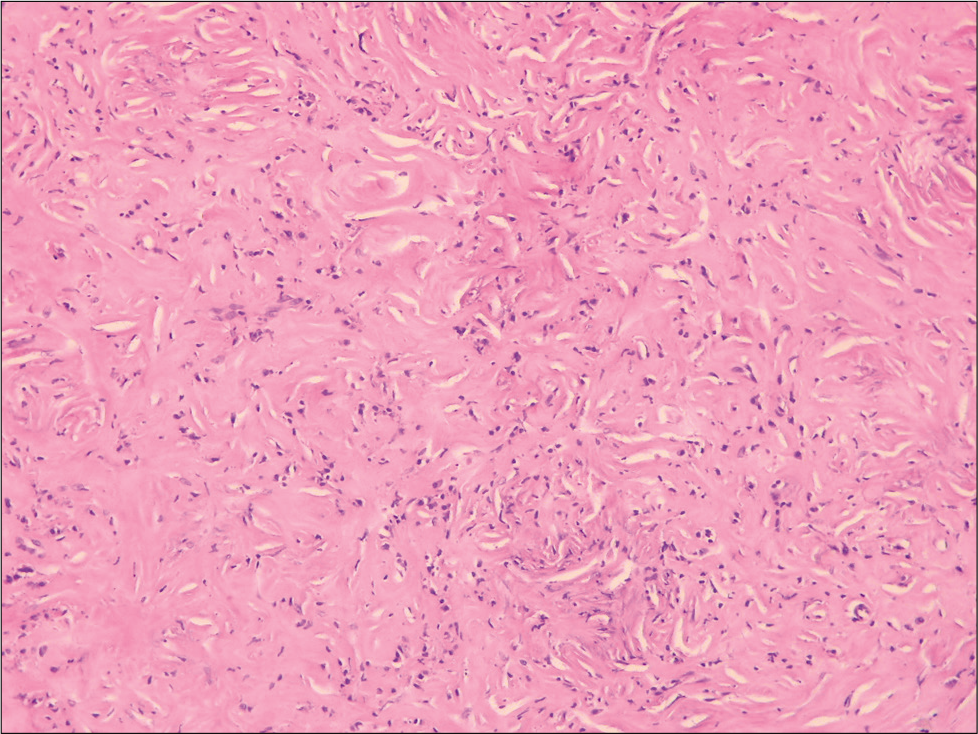
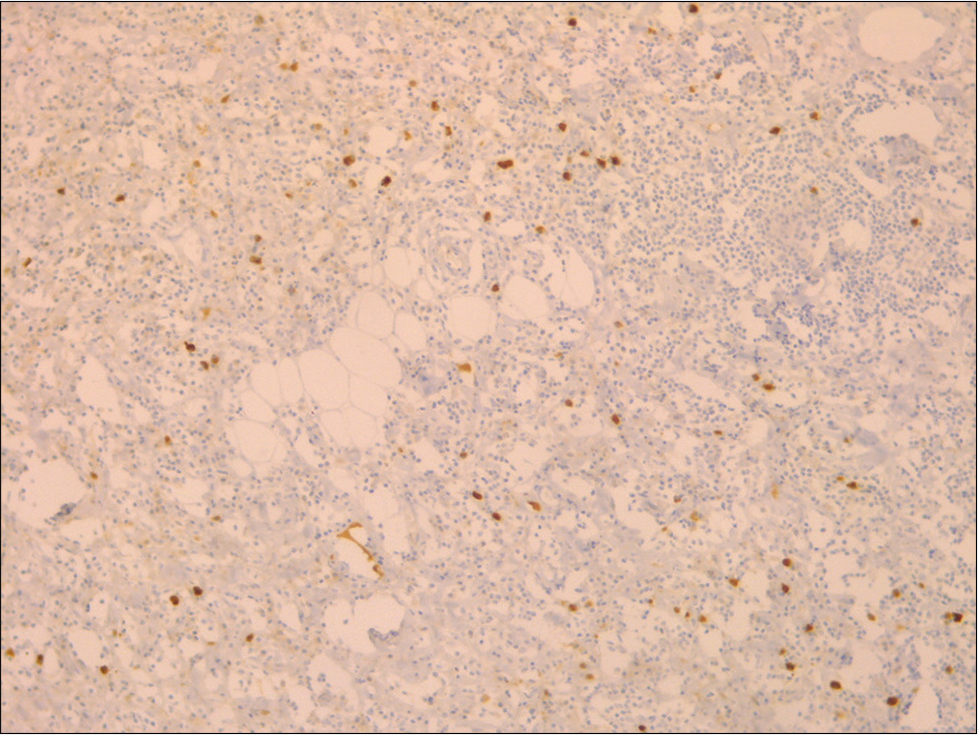
Our patient received high doses of corticosteroids (0.6 mg/kg/day) followed by progressive tapering. His neurological manifestations gradually improved and resolved after 2 months. A cerebral MRI was done 1 month after a well-conducted treatment and showed a reduction of the tumor’s size [
DISCUSSION
In 2001, Hamano et al. were the first to identify IgG4-RD after finding elevated serum levels of IgG4 in patients having autoimmune pancreatitis compared to patients with other causes of chronic pancreatitis.[
The neurological manifestations in IgG4-RD were only documented in <2% of cases and it commonly involves pachymeninges forming hypertrophic pachymeningitis. In a rare instance, it may form inflammatory pseudotumoral masses resembling meningiomas. To the best of our knowledge, only two cases of intracranial spread of IgG4-RD through skull base foramina have been reported in the literature.[
The clinical presentations of IgG4 neuropathy are various. Chronic headache is the most frequent symptom. Patients with IgG4-RD usually have increased serum IgG4 levels. However, this elevation is not constant which makes it a nonspecific index of this pathology.[
Imaging studies in IgG4-RD are useful both for diagnostic and monitoring purposes of these intracranial masses. On computed tomography (CT) scans, lesions are hypodense and demonstrate strong contrast enhancement. On T2-weighted-MRI images, lesions are typically hypointense. T1-weighted images usually demonstrate homogeneous and gradual gadolinium enhancement.[
Despite the increasing frequency of IgG4-related neuropathy, there are no consensus guidelines on its treatment. Commonly, glucocorticoids, usually methylprednisolone or prednisolone, are the first-line treatment with an excellent response.[
When it comes to intracranial pseudotumors, it is still unclear whether a biopsy or partial/subtotal resection is better. Surgical decompression may be required only in the presence of compressive symptoms.[
Finally, the present case suggests that IgG4-RD can form intracranial pseudotumoral masses, meaning that serologic and histologic examinations for IgG4-positive plasma cells must be performed to diagnose the etiology of unusual intracranial masses. However, at present, reports of patients with IgG4-related pseudotumors are still rare. Further reports will be useful to establish this disease concept and guidelines on its treatment.
CONCLUSION
The IgG4-RD is one of the rare differential diagnoses of the central nervous system tumors. Early recognition of IgG4-RD and appropriate establishment of its long-term treatment may avoid unnecessary investigations and morbidity.
Declaration of patient consent
Patient’s consent not required as patient’s identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Baptista B, Casian A, Gunawardena H, D’Cruz D, Rice CM. Neurological manifestations of IgG4-related disease. Curr Treat Options Neurol. 2017. 19: 14
2. Goulam-Houssein S, Grenville JL, Mastrocostas K, Munoz DG, Lin A, Bharatha A. IgG4-related intracranial disease. Neuroradiol J. 2019. 32: 29-35
3. Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001. 344: 732-8
4. Kamisawa T, Okazaki K. Diagnosis and treatment of IgG4-related disease. Curr Top Microbiol Immunol. 2017. 401: 19-33
5. Kamisawa T, Takuma K, Anjiki H, Egawa N, Fujiwara J, Koizumi K. Autoimmune pancreatitis 5 Extra-pancreatic lesions and IgG4-related sclerotic diseases. Nihon Naika Gakkai Zasshi J Jpn Soc Intern Med. 2010. 99: 97-101
6. Katsura M, Mori H, Kunimatsu A, Sasaki H, Abe O, Machida T. Radiological features of IgG4-related disease in the head, neck, and brain. Neuroradiology. 2012. 54: 873-82
7. Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol. 2015. 67: 1688-99
8. Lin CK, Lai DM. IgG4-related intracranial hypertrophic pachymeningitis with skull hyperostosis: A case report. BMC Surg. 2013. 13: 37
9. Lindstrom KM, Cousar JB, Lopes MB. IgG4-related meningeal disease: Clinico-pathological features and proposal for diagnostic criteria. Acta Neuropathol (Berl). 2010. 120: 765-76
10. Lu LX, Della-Torre E, Stone JH, Clark SW. IgG4-related hypertrophic pachymeningitis: Clinical features, diagnostic criteria, and treatment. JAMA Neurol. 2014. 71: 785-93
11. Masaki Y, Shimizu H, Nakamura TS, Nakamura T, Nakajima A, Kawanami HI. IgG4-related disease: Diagnostic methods and therapeutic strategies in Japan. J Clin Exp Hematop. 2014. 54: 95-101
12. Nambirajan A, Sharma MC, Garg K, Sriram S, Boorgula MT, Suri V. Large dural-based mass with bony hyperostosis in a 16-year-old male: IgG4-related disease mimicking lymphoplasmacyte-rich meningioma. Childs Nerv Syst. 2019. 35: 1423-7
13. Perugino CA, Stone JH. Treatment of IgG4-related disease : Current and future approaches. Z Rheumatol. 2016. 75: 681-6
14. Rice CM, Spencer T, Bunea G, Scolding NJ, Sloan P, Nath U. Intracranial spread of IgG4-related disease via skull base foramina. Pract Neurol. 2016. 16: 240-2
15. Tang H, Ding G, Xiong J, Zhu H, Hua L, Xie Q. Clivus inflammatory pseudotumor associated with immunoglobulin G4-related disease. World Neurosurg. 2018. 118: 71-4
16. Toyoda K, Oba H, Kutomi K, Furui S, Oohara A, Mori H. MR imaging of IgG4-related disease in the head and neck and brain. AJNR Am J Neuroradiol. 2012. 33: 2136-9
17. Vlachou PA, Khalili K, Jang HJ, Fischer S, Hirschfield GM, Kim TK. IgG4-related sclerosing disease: Autoimmune pancreatitis and extrapancreatic manifestations. Radiographics. 2011. 31: 1379-402
18. Wallace ZS, Carruthers MN, Khosroshahi A, Carruthers R, Shinagare S, Stemmer-Rachamimov A. IgG4-related disease and hypertrophic pachymeningitis. Medicine (Baltimore). 2013. 92: 206-16

