

- Department of Neurosurgery, Maastricht University Medical Centre, Maastricht, The Netherlands
- Department of Medical Oncology, Maastricht University Medical Centre, Maastricht, The Netherlands
- Department of Pathology, Maastricht University Medical Centre, Maastricht, The Netherlands
- Department of Radiation Oncology, Maastricht University Medical Centre, Maastricht, The Netherlands
- Department of Neurology, Zuyderland Medical Center, Heerlen, The Netherlands
Correspondence Address:
Tim A. M. Bouwens van der Vlis
Department of Neurosurgery, Maastricht University Medical Centre, Maastricht, The Netherlands
DOI:10.4103/sni.sni_166_17
Copyright: © 2017 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Tim A. M. Bouwens van der Vlis, Ann Hoeben, Jan C. Beckervordersandforth, Linda Ackermans, Daniëlle B. P. Eekers, Rianne M. J. Wennekes, Olaf E. M. G. Schijns. Impact of the revised WHO classification of diffuse low-grade glioma on clinical decision making: A case report. 07-Sep-2017;8:223
How to cite this URL: Tim A. M. Bouwens van der Vlis, Ann Hoeben, Jan C. Beckervordersandforth, Linda Ackermans, Daniëlle B. P. Eekers, Rianne M. J. Wennekes, Olaf E. M. G. Schijns. Impact of the revised WHO classification of diffuse low-grade glioma on clinical decision making: A case report. 07-Sep-2017;8:223. Available from: http://surgicalneurologyint.com/surgicalint-articles/impact-of-the-revised-who-classification-of-diffuse-low%e2%80%91grade-glioma-on-clinical-decision-making-a-case-report/
Date of Submission
04-May-2017
Date of Acceptance
02-Jul-2017
Date of Web Publication
07-Sep-2017
Abstract
Background:In the 2016 update of the World Health Organization Classification of Tumors of the central nervous system, phenotypic and genotypic parameters are integrated in diffuse low-grade glioma (LGG) tumor classification. Implementation of this combined phenotypic–genotypic characterization identifies prognostic relevant subgroups.
Case Description:We report a case of a 67-year-old patient with an LGG that showed molecular characteristics similar to glioblastoma multiforme (GBM). After gross total tumor resection, the patient received combination therapy (radiotherapy and chemotherapy) according to high-grade glioma treatment protocol.
Conclusion:The introduction of molecular parameters to the classification of LGG will add a level of objectivity, which will yield biological homogeneous subclasses. Consequently, this will influence patient counseling and clinical decision making regarding treatment protocols.
Keywords: Low-grade glioma, molecular characteristics, treatment, WHO classification
INTRODUCTION
In the 2016 update of the World Health Organization Classification of Tumors of the Central Nervous System phenotypic and genotypic parameters are integrated in diffuse low-grade glioma (LGG) tumor classification. Implementation of this combined phenotypic-genotypic characterization identifies prognostic relevant subgroups
CASE REPORT
A 67-year-old male presented with transient reduced orientation and short-term memory loss. The symptoms lasted for 24 hours during which he also suffered loss of face and object (building) recognition. The patient reported complete amnesia for this episode. Additional amnestic evaluation revealed a period of transient global amnesia of several hours occurring one and a half years ago. His medical history stated diabetes mellitus type II and hypercholesterolemia. Neurological examination at the time of evaluation showed no abnormalities.
Electroencephalography was performed which showed a normal background activity and no signs of epileptic or epileptiform discharges. Magnetic resonance imaging (MRI) showed a nonenhancing lesion with high signal intensity on T2-weighted images and low signal intensity on T1-weighted images of the left mesial temporal lobe [Figure
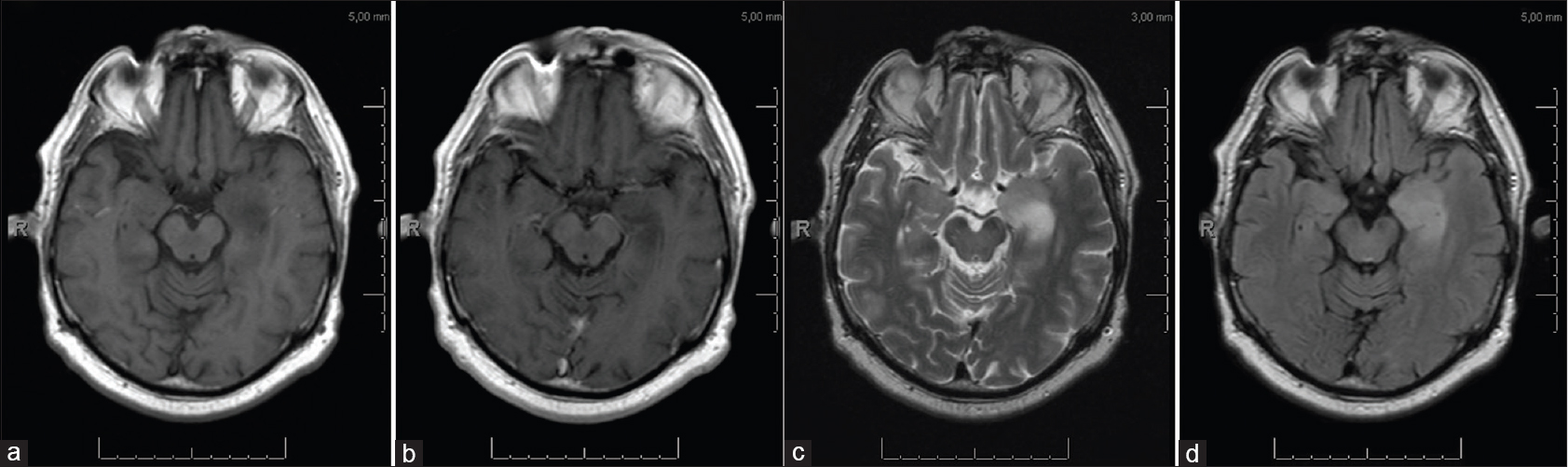
A left-sided anterior temporal lobectomy in combination with resection of the radiologically thickened hippocampus was performed. Intraoperatively, a grayish tumorous tissue of soft consistency was removed suspicious for LGG. A gross total resection was achieved.
A postoperative MRI scan within 72 hours depicted a small area with an increased T2 signal intensity with no abnormal diffusion restrictions, suspicious for residual tumor [Figure
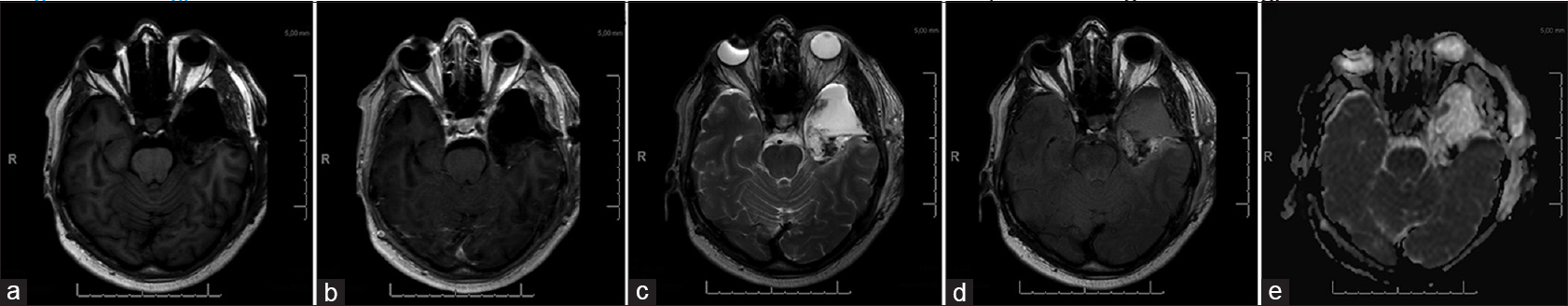
Histology
The obtained tissue showed high cellularity with subpopulations of cells with enlarged nuclei and perinuclear clearing in accordance with a “fried-egg” appearance. The NeuN stain identified multiple neurons. Glial cells stained positive with GFAP, and Ki-67-staining showed an increased proliferative activity in the enlarged atypical cells. The histological diagnosis was an LGG, WHO II, molecularly characterized by the absence of: IDH1/2 mutation, 1p/19q codeletion, or MGMT promotor hypermethylation. See
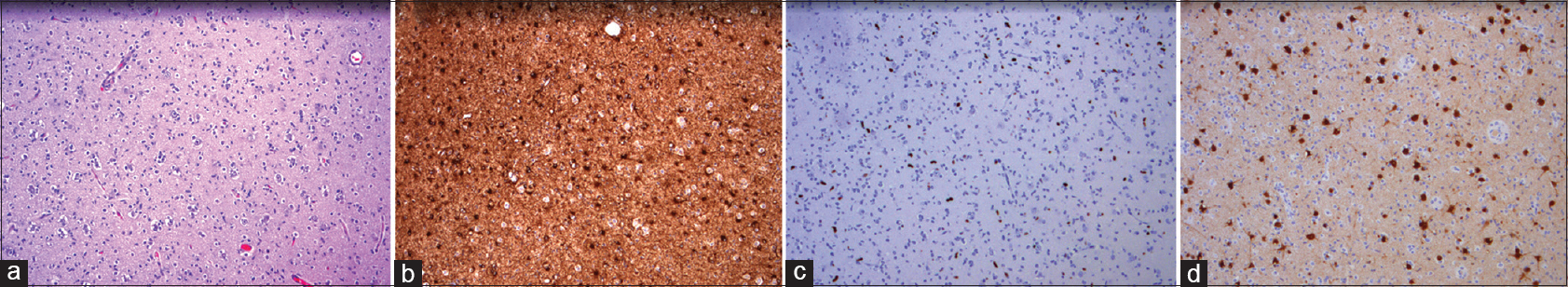
Further genomic analyses using next generation sequencing revealed no mutation in the ATRX, CDKN2a, CIC, FUBP1, NOTCH1, PTEN, and TP53 coding genes and no mutations in the mutational hotspots of BRAF, H3F3A, EGFR, IDH1/IDH2, and PIK3CA. Copy number variation analyses of chromosomes 1, 7, 9, 10, 12, and 19 showed an imbalance of chromosome 7 including EGFR, CD6, and Met with a loss of chromosome 10.
Treatment
The patient was diagnosed according to the 2016 update of the World Health Organization classification of tumors of the central nervous system; IDH wild-type (IDHwt), and diffuse astrocytoma.[
DISCUSSION
Depending on the WHO stage (I–IV), the survival of glioma patients varies from several months to more than 20 years.[
In May 2016, the WHO presented an update of the classification of tumors of the central nervous system.[
LGG and IDH
The first segregation of diffuse LGG is based on the presence of IDH1/2 gene mutation [
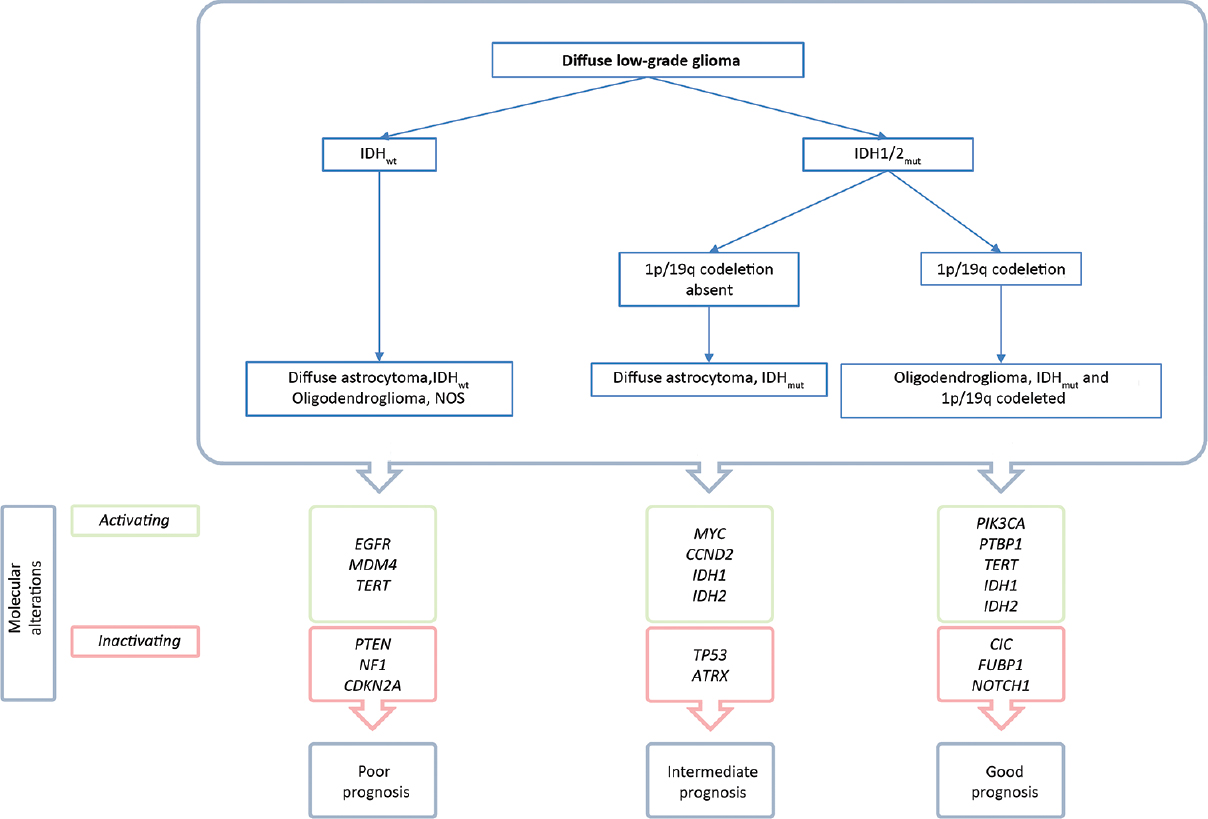
IDH mutations are present in almost 90% of the diffuse LGG and are correlated with a favorable, therapy independent, survival compared to IDHwt LGG: 13.1 years compared to 5.1 years.[
IDHmut with 1p/19q codeletion
Subsequent to IDHmut/IDHwt segregation, LGGs are classified according to the presence or absence of 1p/19q codeletion. A strong correlation exists between the presence an IDH mutation and 1p/19q codeletion with the histological oligodendroglioma with a correspondence rate of 95% for WHO II tumors. Strikingly, mutations in the coding gene for TElomerase Reverse Transcriptase (TERT) is found in 96% of this subclass. TERT gene mutations cause an activation of this enzyme.[
IDHmut without 1p/19q codeletion
Almost all diffuse LGGs without a 1p/19q codeletion harbor mutations in the Tumor protein (TP) 53 coding gene and the majority harbor inactivating mutations in the ATRX gene.[
LGG-IDHwt
LGG with an IDH wild type comprise 10% of all diffuse LGG WHOII tumors. Identification of LGG-IDHwt is of the upmost importance as the majority of this subgroup shows molecular similarities with GBM tumors, with consequent worse prognosis and treatment response.[
CONCLUSION
A subclass of diffuse LGG shows a molecular profile similar to high-grade glioma and is associated with a poor overall survival. Therefore, additional molecular characterization is necessary to identify this subgroup. The 2016 update of the WHO Classification of diffuse LGG facilitates subclass segregation and therefore formally introduces molecular diagnostic results as part of routine neuropathological practice with direct clinical consequences. The presented case illustrates that molecular characterization, beyond the scope of the WHO classification, highly influences adjuvant treatment strategies, and is therefore an example of personalized medicine. Further research will have to show whether the identified subclass-specific genetic aberrations, such as EGFR amplification, will aid to the development of targeted therapy for low-grade gliomas.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med. 2015. 372: 2481-98
2. Buckner JC, Shaw EG, Pugh SL, Chakravarti A, Gilbert MR, Barger GR. Radiation plus Procarbazine, CCNU, and Vincristine in Low-Grade Glioma. N Engl J Med. 2016. 374: 1344-55
3. Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell. 2016. 164: 550-63
4. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009. 462: 739-44
5. Huang FW, Bielski CM, Rinne ML, Hahn WC, Sellers WR, Stegmeier F. TERT promoter mutations and monoallelic activation of TERT in cancer. Oncogenesis. 2015. 4: e176-
6. Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006. 66: 9852-61
7. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016. 131: 803-20
8. Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005. 109: 93-108
9. Pekmezci M, Rice T, Molinaro AM, Walsh KM, Decker PA, Hansen H. Adult infiltrating gliomas with WHO 2016 integrated diagnosis: Additional prognostic roles of ATRX and TERT. Acta Neuropathol. 2017. p.
10. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005. 352: 987-96
11. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012. 483: 479-83
12. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010. 17: 98-110

