

- Department of Neurosurgery, Higashihiroshima Medical Center, Higashihiroshima, Japan
- Department of Neurosurgery, Graduate School of Biomedical Sciences, Hiroshima University, Hiroshima, Japan
Correspondence Address:
Kiyoharu Shimizu
Department of Neurosurgery, Graduate School of Biomedical Sciences, Hiroshima University, Hiroshima, Japan
DOI:10.4103/2152-7806.180765
Copyright: © 2016 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Shimizu K, Yuki K, Sadatomo T, Kurisu K. Isolated neurosarcoidosis presenting with multiple cranial nerve palsies. Surg Neurol Int 19-Apr-2016;7:44
How to cite this URL: Shimizu K, Yuki K, Sadatomo T, Kurisu K. Isolated neurosarcoidosis presenting with multiple cranial nerve palsies. Surg Neurol Int 19-Apr-2016;7:44. Available from: http://surgicalneurologyint.com/surgicalint_articles/isolated-neurosarcoidosis-presenting-with-multiple-cranial-nerve-palsies/
Abstract
Background:As an extremely rare subtype of sarcoidosis that develops exclusively in the nervous system, isolated neurosarcoidosis is difficult to diagnose. In addition, its exact clinical features are not known.
Case Description:A 61-year-old man presented with right ear hearing loss, diplopia, and fever. Computed tomography (CT) and magnetic resonance imaging revealed mass lesions in the right cerebellum and left side body of the lateral ventricle. Neither systemic CT nor positron emission tomography revealed extracranial lesions. A neuroendoscopic biopsy was performed on the lateral ventricle lesion, and a histopathology analysis revealed epithelioid granulomatous inflammation. By systematic exclusion of other possible granulomatous diseases, isolated neurosarcoidosis was diagnosed. The lesions disappeared immediately upon corticosteroid (methylprednisolone) treatment and had not recurred as of a 12-month follow-up examination.
Conclusions:Isolated neurosarcoidosis is difficult to diagnose. Successful diagnosis requires compatible clinical findings, histological demonstration of noncaseating granulomas, and exclusion of other granulomatous diseases. Isolated neurosarcoidosis has a relatively good clinical prognosis, which could be characteristic of the disease.
Keywords: Cranial nerve palsy, granulomatous inflammation, isolated neurosarcoidosis
INTRODUCTION
Sarcoidosis is a multisystem granulomatous disease of unknown etiology. Sarcoidosis can occur in the central or peripheral nervous system with other organ involvement (neurosarcoidosis), however, when sarcoidosis develops exclusively in the nervous system, it is called isolated neurosarcoidosis. Although its precise prevalence has not been determined, isolated neurosarcoidosis is estimated to account for <1% of sarcoidosis cases.[
CASE REPORT
A 61-year-old man with a history of hypertensive cerebellar hemorrhage visited our hospital because of diplopia and fever. The diplopia was recognized when he gazed at the right side and disappeared with monocular vision. In addition, he became aware of right-ear hearing loss a few months before visiting the hospital. He was alert and had no paralysis. The deep tendon reflexes were normal, and any upper motor neuron pathological reflexes were not seen. His tandem walk was disturbed and leaned to the right side. An ocular examination showed faint right lateral gaze palsy. Fundus examination revealed papilledema. Laboratory results included the following: C-reactive protein 0.01 mg/dL (0–0.3 mg/dL), lactate dehydrogenase 142 U/L (119–229 U/L), erythrocyte sedimentation rate = 24 mm/1 h (0–10 mm/1 h), albumin = 3.8 g/dL (4.0–5.0 g/dL), calcium = 8.6 mg/dL (8.6–10.1 mg/dL), angiotensin conversion enzyme = 7.7 IU/L (8.3–21.4 IU/L), soluble interleukin 2 receptor = 291 U/ml (145–519 U/ml), and immunoglobulin G4 = 40.6 mg/dL (4.8–105 mg/dL). Antinuclear antibody test was negative. QuantiFERON® blood tests were negative, and tests for Epstein-Barr virus, cytomegalovirus, toxoplasma, syphilis, hepatitis B virus, hepatitis C virus, and human immunodeficiency virus were all negative. Cerebrospinal fluid (CSF) study revealed a lymphocytosis and elevated protein levels: Cell count 349/mm3 (pleocyte 16/mm3, lymphocyte 333/mm3) and total protein 153 mg/dL. Oligoclonal band was negative.
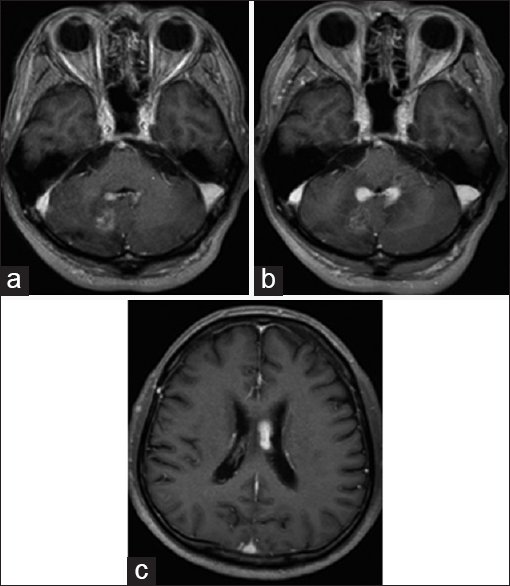
The chest X-ray was normal and no bilateral hilar lymphadenopathy was seen. Magnetic resonance imaging (MRI) revealed intracranial mass lesions in the right hemisphere of the cerebellum and the left body of the lateral ventricle [
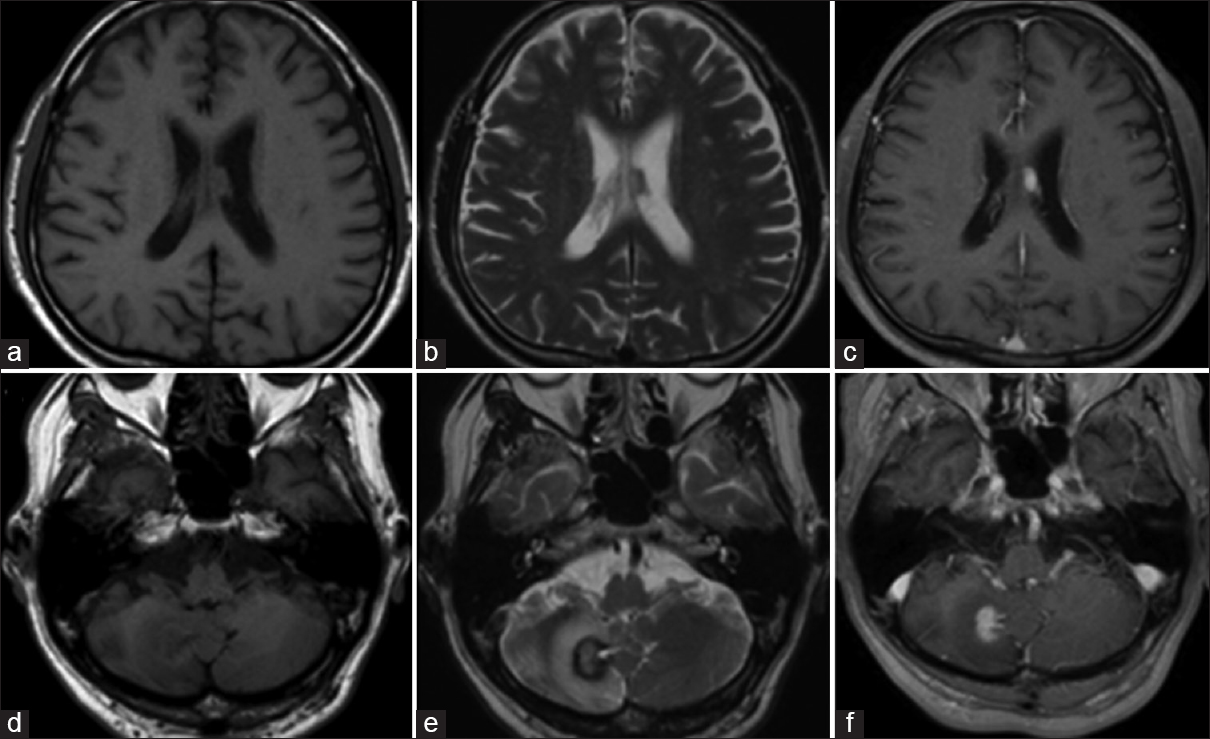
Figure 1
Magnetic resonance imaging on admission. The two mass lesions in the left body of the lateral ventricle and the right cerebellum hemisphere were isointense on T1-weighted images (a and d), and hypointense on T2-weighted images (b and e). The lesions were enhanced after gadolinium-diethylenetriamine pentaacetate injection (c and f)
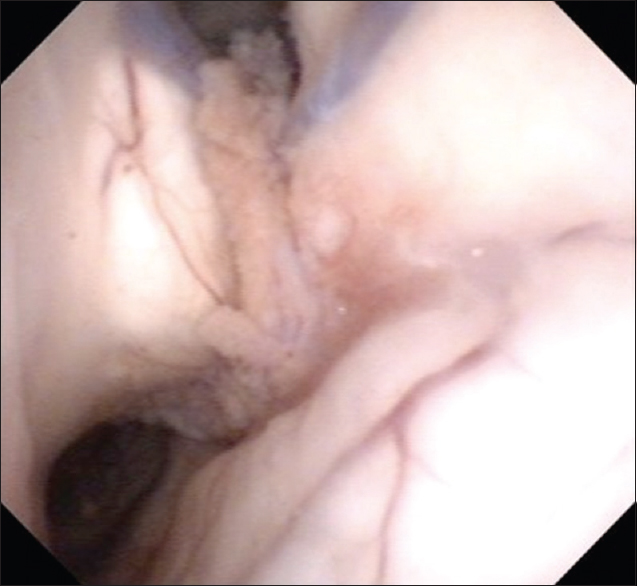
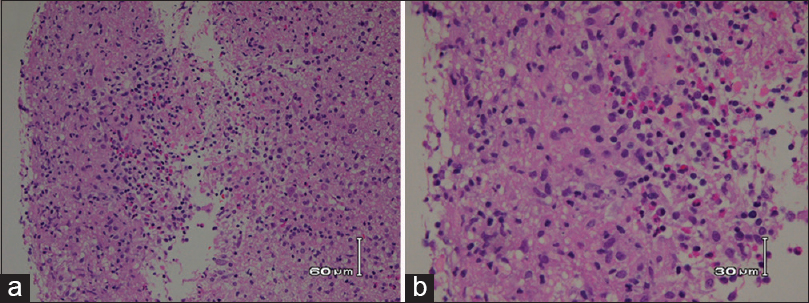
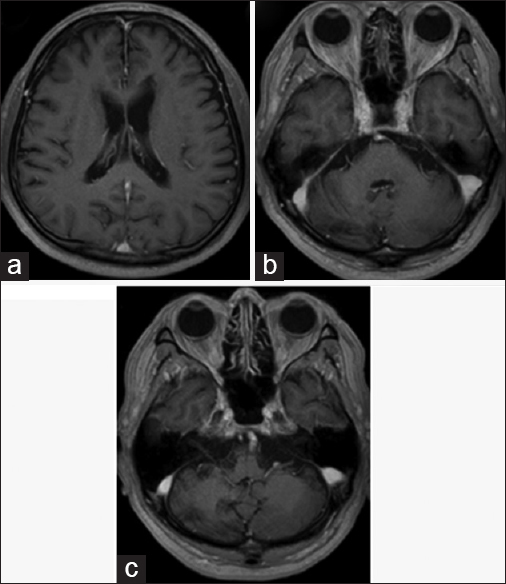
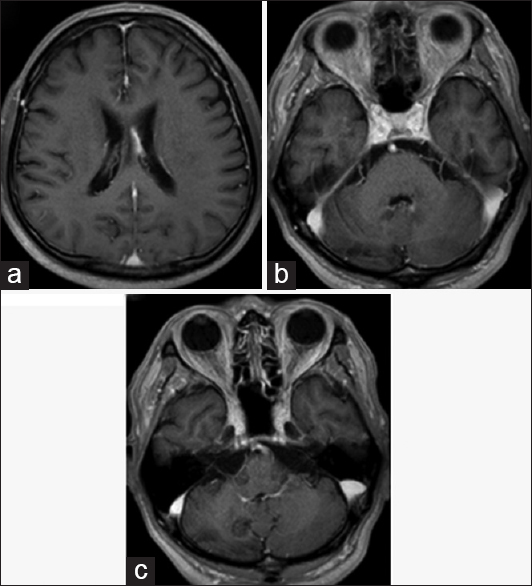
DISCUSSION
Isolated neurosarcoidosis is an extremely rare disease, and its accurate diagnosis is difficult. The accurate diagnosis of isolated neurosarcoidosis requires compatible clinical symptoms, a histological finding of noncaseous granulomatous inflammation, and exclusion of other granulomatous diseases.[
An accurate diagnosis of isolated neurosarcoidosis requires characteristic clinical presentations, the presence of granulomatous inflammation on tissue biopsy, and exclusion of other possible diagnoses. Neurosarcoidosis could occur in any part of the nervous systems; however, the basal leptomeninges, the hypothalamus, and the pituitary gland are most commonly involved.[
Isolated neurosarcoidosis tends to have a more favorable clinical prognosis than does neurosarcoidosis with extracranial organ involvement.[
The diagnosis of isolated neurosarcoidosis requires compatible clinical findings, histological demonstration of noncaseating granulomas, and exclusion of other granulomatous diseases. In the present case study, multiple cranial nerve palsies, a relapsing and remitting clinical course, and granulomatous inflammation in the tissue biopsy contributed to the diagnosis. Isolated neurosarcoidosis tends to have more favorable clinical outcomes than neurosarcoidosis with extracranial organ involvement. Such a good prognosis was experienced by our patient, and this information could be useful to clinicians seeking to mitigate stress-related complications in patients anxious over the symptoms they are experiencing.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Agbogu BN, Stern BJ, Sewell C, Yang G. Therapeutic considerations in patients with refractory neurosarcoidosis. Arch Neurol. 1995. 52: 875-9
2. Brinar VV, Habek M. Isolated central nervous system sarcoidosis: A great mimicker. Clin Neurol Neurosurg. 2008. 110: 939-42
3. Bromberg JE, Siemers MD, Taphoorn MJ. Is a “vanishing tumor” always a lymphoma?. Neurology. 2002. 59: 762-4
4. Ferriby D, de Seze J, Stojkovic T, Hachulla E, Wallaert B, Destée A. Long-term follow-up of neurosarcoidosis. Neurology. 2001. 57: 927-9
5. Gullapalli D, Phillips LH. Neurologic manifestations of sarcoidosis. Neurol Clin. 2002. 20: 59-83, vi
6. Hoitsma E, Faber CG, Drent M, Sharma OP. Neurosarcoidosis: A clinical Dilemma. Lancet Neurol. 2004. 3: 397-407
7. Joseph FG, Scolding NJ. Neurosarcoidosis: A study of 30 new cases. J Neurol Neurosurg Psychiatry. 2009. 80: 297-304
8. Judson MA. The diagnosis of sarcoidosis. Clin Chest Med. 2008. 29: 415-27, viii
9. Kellinghaus C, Schilling M, Lüdemann P. Neurosarcoidosis: Clinical experience and diagnostic pitfalls. Eur Neurol. 2004. 51: 84-8
10. Loor RG, van Tongeren J, Derks W. Multiple cranial nerve dysfunction caused by neurosarcoidosis. Am J Otolaryngol. 2012. 33: 484-6
11. Nowak DA, Widenka DC. Neurosarcoidosis: A review of its intracranial manifestation. J Neurol. 2001. 248: 363-72
12. Nozaki K, Scott TF, Sohn M, Judson MA. Isolated neurosarcoidosis: Case series in 2 sarcoidosis centers. Neurologist. 2012. 18: 373-7
13. Oh JH, Park K, Lee SJ, Shin YR, Choung YH. Bilateral versus unilateral sudden sensorineural hearing loss. Otolaryngol Head Neck Surg. 2007. 136: 87-91
14. Okita Y, Narita Y, Miyakita Y, Ohno M, Fukushima S, Maeshima A. Long-term follow-up of vanishing tumors in the brain: How should a lesion mimicking primary CNS lymphoma be managed?. Clin Neurol Neurosurg. 2012. 114: 1217-21
15. Peltomaa M, Pyykkö I, Sappälä I, Viitanen L, Viljanen M. Lyme borreliosis, an etiological factor in sensorineural hearing loss?. Eur Arch Otorhinolaryngol. 2000. 257: 317-22
16. Riku Y, Ito M, Atsuta N, Watanabe H, Momota H, Sobue G. Intracranial germinoma masquerading as a granulomatous inflammation, diagnostic failure after brain biopsy. Rinsho Shinkeigaku. 2013. 53: 835-8
17. Scott TF, Yandora K, Kunschner LJ, Schramke C. Neurosarcoidosis mimicry of multiple sclerosis: Clinical, laboratory, and imaging characteristics. Neurologist. 2010. 16: 386-9
18. Smith JK, Matheus MG, Castillo M. Imaging manifestations of neurosarcoidosis. AJR Am J Roentgenol. 2004. 182: 289-95
19. . Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med. 1999. 160: 736-55

