

- Department of Neuropathology, Children’s Hospital of Los Angeles, University of Rochester School of Medicine, Rochester, United States
- Department of Neurosurgery, Children’s Hospital of Los Angeles, University of Rochester School of Medicine, Rochester, United States
- Department of Hematology/Oncology, Children’s Hospital of Los Angeles, University of Rochester School of Medicine, Rochester, United States
- Department of Imaging sciences, Children’s Hospital of Los Angeles, University of Rochester School of Medicine, Rochester, United States
- Department of Pathology, Children’s Hospital of Los Angeles, University of Rochester School of Medicine, Rochester, United States.
Correspondence Address:
Mahlon D. Johnson, MD, PhD, Professor and Director of Neuropathology, University of Rochester School of Medicine, Rochester, United States.
DOI:10.25259/SNI_830_2023
Copyright: © 2024 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Mahlon Johnson1, Howard Silberstein2, David Korones3, Ali Hussain4, Debra Hawes5. Low-grade fibromyxoid tumor of the dura: A new entity?. 19-Jan-2024;15:14
How to cite this URL: Mahlon Johnson1, Howard Silberstein2, David Korones3, Ali Hussain4, Debra Hawes5. Low-grade fibromyxoid tumor of the dura: A new entity?. 19-Jan-2024;15:14. Available from: https://surgicalneurologyint.com/surgicalint-articles/12716/
Date of Submission
07-Oct-2023
Date of Acceptance
22-Nov-2023
Date of Web Publication
19-Jan-2024
Abstract
Background: Low-grade fibromyxoid tumors are uncommon in children. Their differentiation from high-grade fibromyxoid tumors, as seen in adults, is imperative to diagnosis. Awareness of the entity and its subsequent behavior may guide management and predict outcomes.
Case Description: We describe the case of a previously unreported low-grade fibromyxoid tumor of the cerebellum in an 8-year-old male. Extensive immunohistochemical, next-generation sequencing, and attempted DNA methylation profiling are reported. There has been no recurrence during the 6-year follow-up. Screening excluded multiple myxoid tumors, including low-grade fibromyxoid sarcoma. The findings suggest that, with gross total resection, the lesions may not recur.
Conclusion: The case of fibromyxoid tumor with 6-year follow-up and the limited literature of similar tumors are reviewed.
Keywords: Dura, Fibromyxoid, Meningioma
INTRODUCTION
Dural-based neoplasms are relatively uncommon in children, and fibromyxoid dural tumors are even rarer.[
CASE DESCRIPTION
An 8-year-old male presented after two weeks of progressively worsening headaches, nausea, and vomiting. Before that, he had fallen, hitting his head on cement. On examination, his speech was intact, and he had no visual changes, facial droop, weakness, issues with memory loss, or altered mental status. There had been no changes in his sense of smell, hearing, or taste.
He was well in school and had been free of any fever, cough, congestion, sore throat, chest pain, diarrhea, dysuria, difficulty breathing, neck pain, or pain elsewhere. There had been no weight loss.
The family history included pancreatic cancer in a maternal grandfather and breast cancer in a maternal grandmother. However, a teenage first cousin had a brain tumor.
The patient had no significant past medical history. He had four healthy siblings.
On examination, he had a blood pressure of 92/42 mmHg, pulse 52, and temperature of 37.2°C. No lymphadenopathy was found. The neurologic examination revealed mild truncal ataxia, an abnormal tandem gait, and a heel-shin test on the left side. His laboratory analyses revealed a normal but slightly elevated lactate dehydrogenase . An initial computed tomography scan revealed a complex multiloculated cystic mass centered within the left cerebellar hemisphere, with moderate mass effect and partial effacement of the fourth ventricle. There was minimal dilatation of the supratentorial ventricular system. Magnetic resonance imaging (MRI) revealed a large, well-circumscribed mass centered within the left cerebellar hemisphere, with apparent exophytic extension through the posterolateral cortical margin and suggested adherence to the overlying endosteal dural layer [
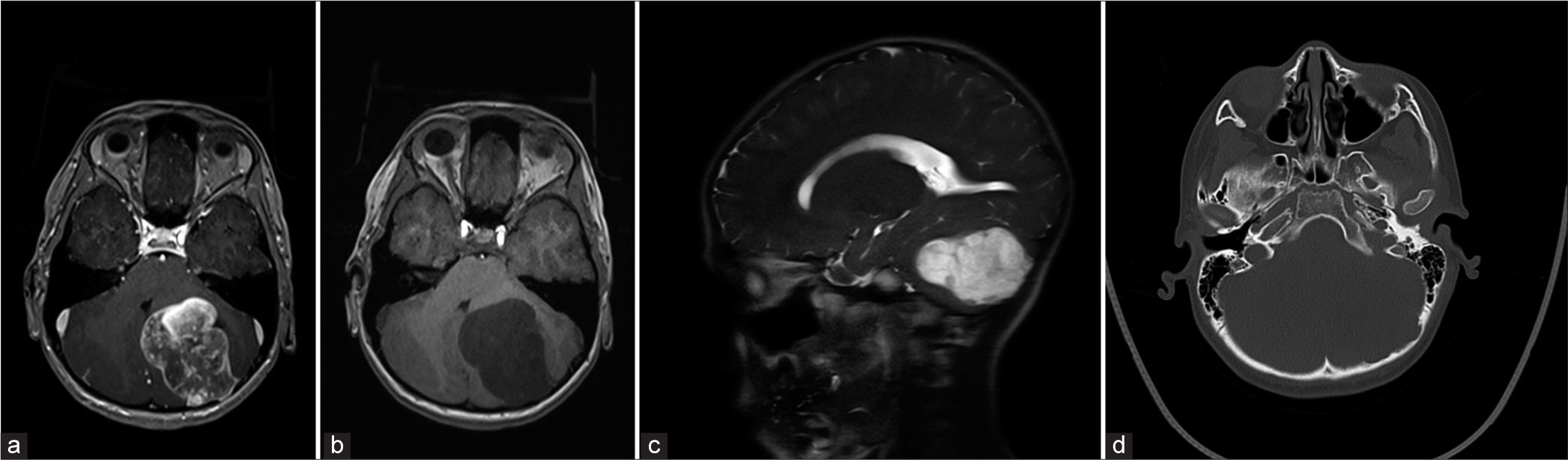
Figure 1:
(a) MRI axial post gadolinium axial T1 weighted image. The tumor was a large well circumscribed, dura based, extra-axial mass in the left aspect of the posterior fossa. (b) The mass is predominantly hypointense on T1 weighted image. (c) The mass lesion showed marked hyperintensity on T2 weighted images. (d) Remodeling and scalloping of the inner table of the occipital bone was also seen.
Clinically, a diagnosis of atypical pilocytic astrocytoma was favored. However, the tumor was heterogeneous on imaging, leading to a consideration of other possible diagnoses, such as a medulloblastoma or a malignant glioma.
At surgery, the left-sided intradural, extra-axial tumor was found to have eroded through the dura with a consistency of an extra-axial meningioma. Gross total resection was achieved. MRIs 9 months after surgery revealed no abnormal enhancement or evidence of recurrence.
The sections revealed a tumor with large hypocellular and moderately cellular areas with scattered stellate and spindle cells.
The toluidine blue stain revealed scattered mast cells. Alcian blue staining was extensive. The periodic acid schiff (PAS) showed no loss with periodic acid schiff, diastase (PASD). Stellate and spindle cells showed scattered epithelial membrane antigen (EMA), S100, rare progesterone receptor, no pan-cytokeratin, no CK18, and no calretinin immunoreactivity. There was neurofilament immunoreactivity in one of two blocks, and focally, collagen bundles were found, but no notable collagen IV immunoreactivity was found. No ALK1, CD1a, or CD117 immunoreactivity was seen. CD68 and CD163 highlighted macrophages [
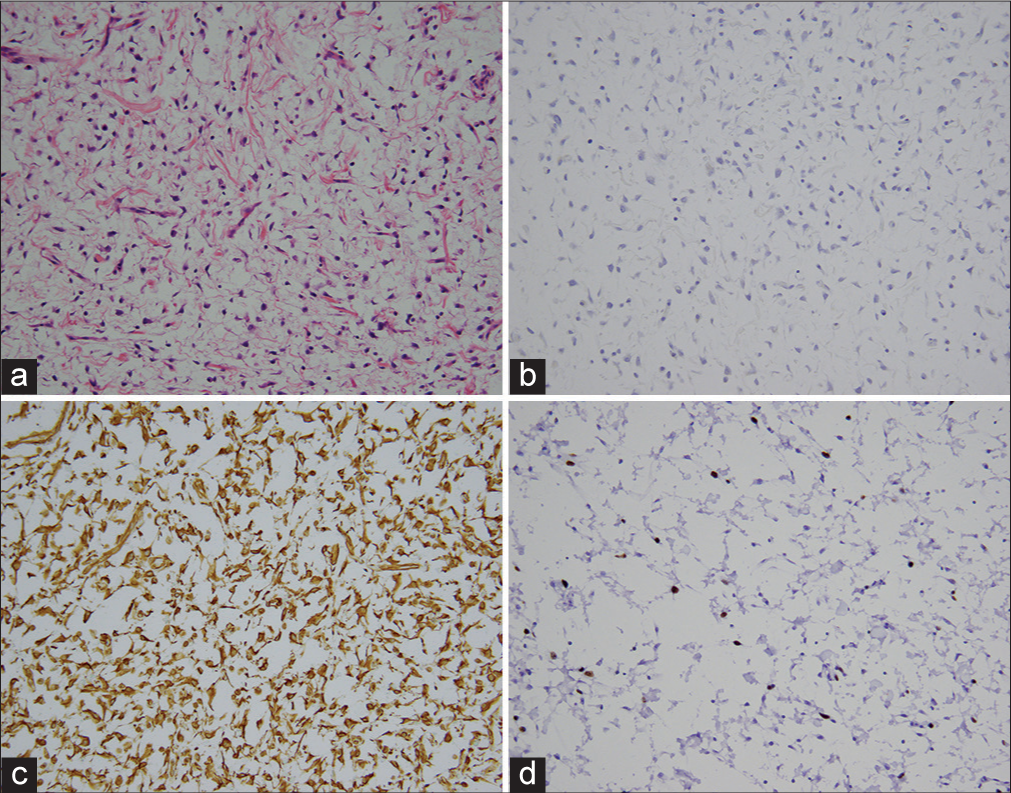
Figure 2:
(a) Myxoid tumor with spindle cells. (Hematoxylin and eosin, original magnification × 200). (b) Epithelial membrane (EMA) immunohistochemistry. No brown EMA immunoreactivity was detected in spindle cells. (Hematoxylin counterstain and diaminobenzidine chromogen (brown); original magnification × 200). (c) Vimentin immunohistochemistry. Tumor cells displayed extensive vimentin immunoreactivity (brown). (Hematoxylin counterstain and diaminobenzidine; original magnification × 200). (d) Ki-67 immunohistochemistry. Ki-67 labeling (brown) was variable (Hematoxylin counterstain and diaminobenzidine; original magnification × 200).
Vimentin immunostaining was extensive. Beta-catenin was cytoplasmic. Langerin was found in scattered cells.
In some areas, several mitoses were found, and the Ki-67 labeling is focused at approximately 13% or higher [
Tumor cells showed no immunoreactivity with SMA, MSA, synaptophysin, glial fibrillary acidic protein, desmin, and CAM 5.2. CD34 was confined largely to blood vessels. Fluorescence in situ hybridization for FUS (16p11.2) revealed no rearrangement of the gene. Subsequently, DNA sequencing revealed no mutations in AKT1, ALK, BRAF, CDK4, CTTNB1, DDDR2, EGFR, ERB2-4, FGFR12 and 3, IGH1 and 2, JAK2 and 3, KIT, KRAS, PAM2K1 and 2, MET, MTOR, PDGFRA, PIK3CA, RAF1, PET, ROS, and SMO genes. Genome-wide DNA methylation at Knight Diagnostic laboratory, attempted multiple times on two different tissue blocks, was unsuccessful due to low DNA concentration. The tumor was classified as a low-grade fibromyxoid lesion.
Six years after surgery, the patient has had no recurrence. He is clinically well and active in sports, requiring balance and fine motor skills.
DISCUSSION
Fibromyxoid tumors must be differentiated from a number of fibrous and myxoid lesions including fibromas,[
The age, absence of identifiable areas of classic meningioma, and lack of expression of EMA and progesterone receptor immunoreactivity argue against a myxoid meningioma. The lesion does appear to be fibromatous, but the lack of cellularity and cellular atypia would be unusual for a meningeal fibroma. The lesion has some features suggestive of myxo-inflammatory fibroblastic sarcoma, including atypical binucleated cells and inflammatory infiltrates. Nonetheless, this is a rare tumor that usually arises in soft tissue in acral sites and shows a FUS gene rearrangement.[
The low-grade fibromyxoid tumor here was a hypocellular lesion composed of rare cells with elongated nuclei, dense collagen with no mitotic activity, and notable alcian blue staining highlighting the myxoid component. The chromosomal analysis did not identify additional features associated with neoplasia. In our case, the designation fibromyxoid tumor was tentatively descriptive and suggestive of a tumor with a low risk of recurrence and aggressive growth. Follow-up for six years supported this. The designation is not meant to imply that this is a variant of a low-grade fibromyxoid sarcoma. The latter tumors are distinct from this tumor by age and location, as well as lack of FUS gene region rearrangement. In addition, our tumor was different from the myxoinflammatory fibroblastic sarcomas’ tendency to occur in the distal extremities of adults.[
CONCLUSION
The classification and pathogenesis of this and similar tumors remain to be established. In the absence of other descriptions of similar tumors, it may represent a new entity different from fibromyxoid tumors in adults. Given the favorable, essentially curative outcome with surgery alone, awareness of its behavior is important.
Statement of ethics
Written consent was obtained from the parents for publication of the details of the medical case and images.
Author’s contributions
MDJ made the diagnosis and wrote the manuscript (MS). HS performed the surgery and contributed to the MS. DK contributed to the MS. AH provided images and contributed to the MS. DH performed the molecular analysis and contributed to the MS.
Ethical approval
Institutional Review Board approval is not required.
Data availability
There are no datasets. All information is in the MS.
Declaration of patient consent
Patient’s consent not required as patient’s identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
1. Antonescu C, Ladanyi M, Christopher DM, Fletcher K, Unni K, Mertens F, editors. Myxoid liposarcomas. Tumours of soft tissue and bone. Lyon France: IARC Press; 2002. p. 40-3
2. Graham JF, Loo SY, Matoba A. Primary brain myxoma, an unusual tumor of meningeal origin: Case report. Neurosurgery. 1999. 45: 166-9 discussion 169-70
3. Harrison JD, Rose PE. Myxoid meningioma: Histochemistry and electron microscopy. Acta Neuropathol (Berl). 1985. 68: 80-2
4. Ieremia E, Thway K. Myxoinflammatory fibroblastic sarcoma. Arch Pathol Lab Med. 2014. 138: 1406-11
5. Johnson MD, Hussain A. Imaging and clinicopathologic features of myxoid meningiomas. Clin Neuropathol. 2019. 38: 238-44
6. Johnson MD, Powell SZ, Boyer PJ, Weil RJ, Moots PL. Dural lesions mimicking meningiomas. Human Pathol. 2002. 33: 1211-26
7. Kleinschmidt-DeMasters BK, Boovier C, Hainfellner JA, Perrt A, editors. Intracranial mesenchymal tumour, FET: CREB fusion positive. Pathology and genetics, tumours of the nervous system. World Health Organization classification of tumours. Lyon, France: WHO Classification of Tumours Editorial Board, IARC Press; 2021. p. 317-9
8. Krisht KM, Altay T, Couldwell WT. Myxoid meningioma: A rare metaplastic meningioma variant in a patient presenting with intratumoral hemorrhage. J Neurosurg. 2012. 116: 861-5
9. Medeiros F, Scheithauer BW, Oliveria AM, Gregory RS. Angiomyxofirbomatous tumor of the falx cerebri. Am J Surg Pathol. 2006. 30: 545-7
10. Pollack LF, Hamilton RL, Fitz C, Orenstein DM. An intrasylvian “fibroma” in a child with cystic fibrosis: Case report. Neurosurgery. 2000. 46: 744-7
11. Sahm F, Brastianos PK, Claus EB, Mawrin C, Perry A, Santagata S, editors. Meningioma variants. Pathology and genetics, tumours of the nervous system. World Health Organization classification of tumours. Lyon, France: WHO Classification of Tumours Editorial Board, IARC Press; 2021. p. 237-45
12. Van Roggan G, Hogendoorn PC, Fletcher CD. Myxoid tumors of soft tissue. Histopathology. 1999. 35: 291-312

