

- Department of Neurosurgery, Dharmais National Cancer Hospital, Jakarta, Indonesia
- Department of Plastic Surgery, Dharmais National Cancer Hospital, Jakarta, Indonesia
- Department of Neurology, Dharmais National Cancer Hospital, Jakarta, Indonesia
- Department of Pathology Anatomy, Dharmais National Cancer Hospital, Jakarta, Indonesia
- Department of Neurosurgery and Oncology & Stem Cell Working Group, Universitas Padjadjaran, Bandung, Indonesia
Correspondence Address:
Arwinder Singh Gill
Department of Neurosurgery and Oncology & Stem Cell Working Group, Universitas Padjadjaran, Bandung, Indonesia
DOI:10.4103/sni.sni_196_17
Copyright: © 2018 Surgical Neurology International This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.How to cite this article: Muhammad Firdaus, Arwinder Singh Gill, Dewi Aisiyah Mukarramah, Rini Andriani, Lenny Sari, Dian Cahyanti, Ahmad Faried. Malignant peripheral nerve sheath tumor of the scalp: Two rare case reports. 15-May-2018;9:102
How to cite this URL: Muhammad Firdaus, Arwinder Singh Gill, Dewi Aisiyah Mukarramah, Rini Andriani, Lenny Sari, Dian Cahyanti, Ahmad Faried. Malignant peripheral nerve sheath tumor of the scalp: Two rare case reports. 15-May-2018;9:102. Available from: http://surgicalneurologyint.com/surgicalint-articles/malignant-peripheral-nerve-sheath-tumor-of-the-scalp-two-rare-case-reports/
Date of Submission
29-May-2017
Date of Acceptance
12-Apr-2018
Date of Web Publication
15-May-2018
Abstract
Background:Malignant peripheral nerve sheath tumors (MPNSTs) constitute a group of soft tissue neoplasm with neuroectodermal origin. Most cases are small at presentation and only some have been described reaching giant dimensions.
Case Description:We report two cases that were diagnosed and treated as giant MPNST of the scalp. Both patients had extensive lesion on the head with intracranial infiltration. Microsurgical resection was indicated and a vascularized free flap was used to cover the defect. During follow-up the tumors recurred and further surgical excision treatment by adjuvant radiation therapy was performed.
Conclusion:MPNSTs of the scalp are rare neoplasm of the peripheral nervous system. They are aggressive lesion that can recur and their management requires a multimodality approach.
Keywords: Malignant peripheral nerve sheath tumors, scalp, soft tissue tumor
INTRODUCTION
Malignant peripheral nerve sheath tumors (MPNSTs) of the scalp are rare neoplasms of the nervous system. These tumors are considered to be a subcategory of soft tissue sarcomas based on World Health Organization (WHO) classification on central nervous system (CNS) tumors,[
MPNSTs are very rare tumors with incidents of approximately 0.001% in the population;[
In this report, we present two cases adding the evidence of scalp MPNST in world literature that were diagnosed and treated as giant MPNST of the scalp. Patients underwent microsurgery and adjuvant radiotherapy.
CASE REPORTS
Case 1
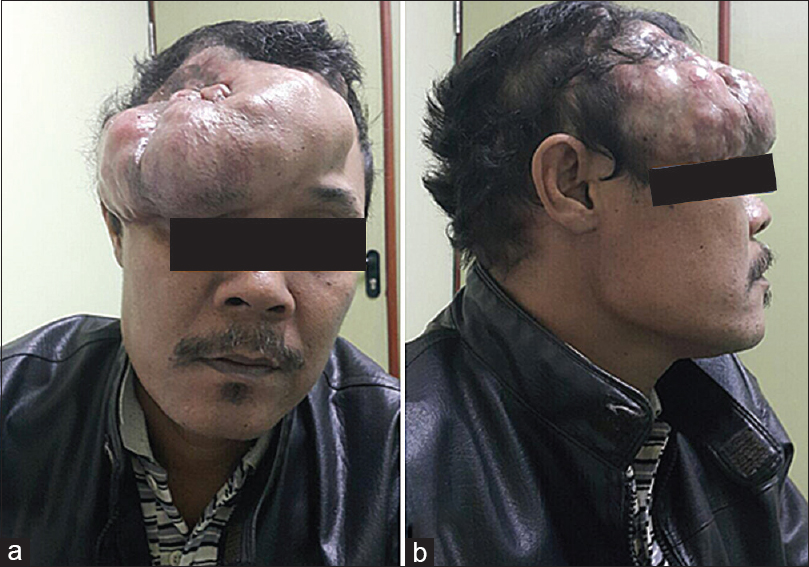
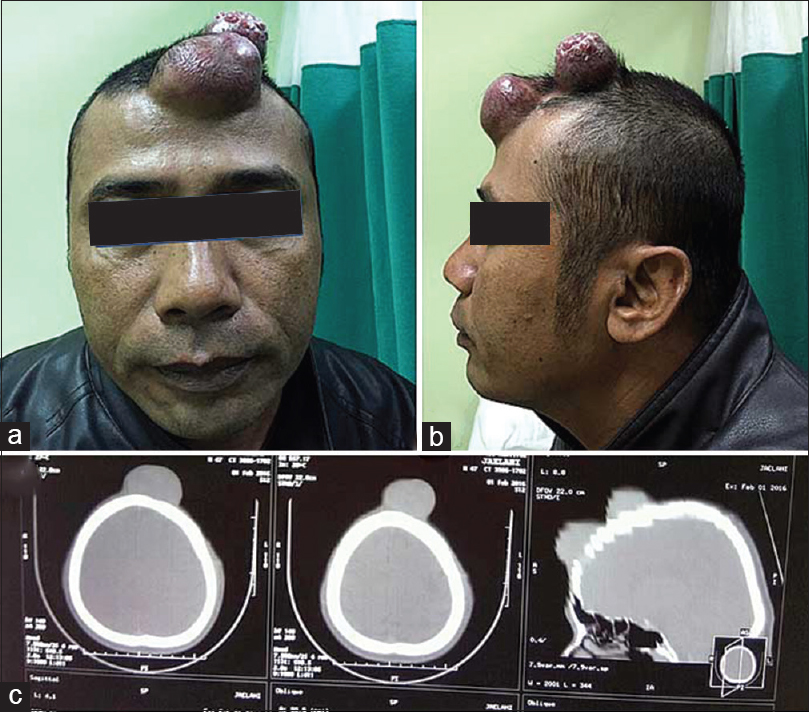
A 45-year-old male presented with painless and progressive swelling on the right frontal region over 2 years prior to examination. According to patient history, the “swelling” started over his eyebrow and progressively increased in size. He underwent a biopsy at an outside facility and the pathology report diagnosed neurofibroma but patient failed to follow-up. He was later referred to our hospital after the lesion enlarged in size and he had developed difficulties opening his eyes. He had no history of trauma, bone pain, systemic disease, or neurological symptoms. Physical examination found an extensive scalp lesion that measured 30 × 20 cm in size, extending from the right orbital rim toward the contralateral side and the parietal region. Upon palpation the lesion was firm but without any tenderness [
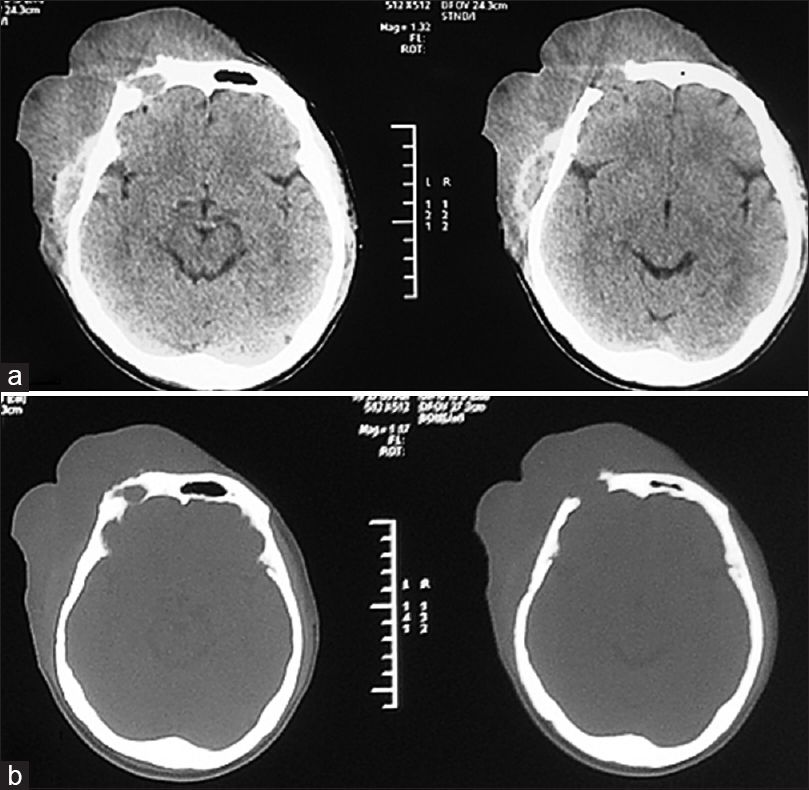
Computed tomography (CT) scan of the head revealed a calvarial soft tissue mass predominantly located in right frontoparietal with infiltrating mass on the frontal region and associated right frontal bone defect [Figure
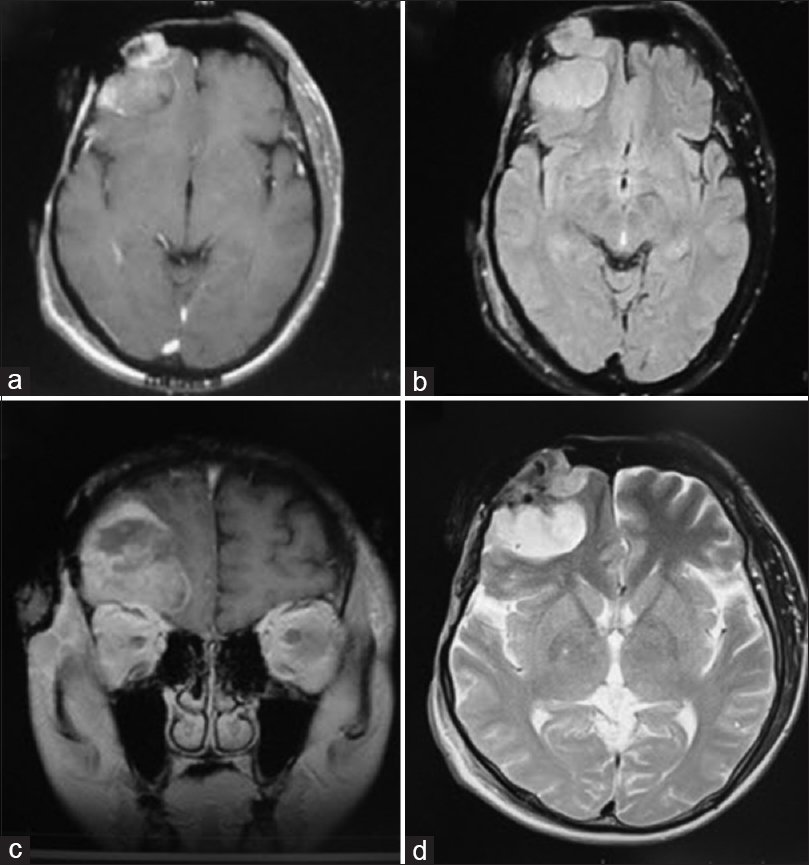
The patient was positioned supine with back slightly elevated 20° and without any head fixation. A wide marginal excision with 4 cm distance from neoplasm margin was performed. At surgery, tumor tissue was found to be soft, fleshy, moderately vascular, and mostly encapsulated with some areas displaying ill-defined margins. The mass was eroding through the internal table of the bone and infiltrated the dura mater as well as the intradural compartment. The mass was highly vascular and bled easily when touched. The bone at the right frontal region appeared moth-eaten and was removed with rongeurs until a normal hard and thick border was identified. A wide intracranial portion of the lesion was removed without any involvement of brain parenchyma, and a fascia lata graft was used for duroplasty. The postoperative bone defect measuring 10 × 10 cm was closed using a titanium mesh. A vascularized free flap was raised from anterolateral thigh and sewn in by the plastic surgeon to close the skin defect. The patient's neurological status remained intact postoperatively. He was discharged from the hospital uneventfully.
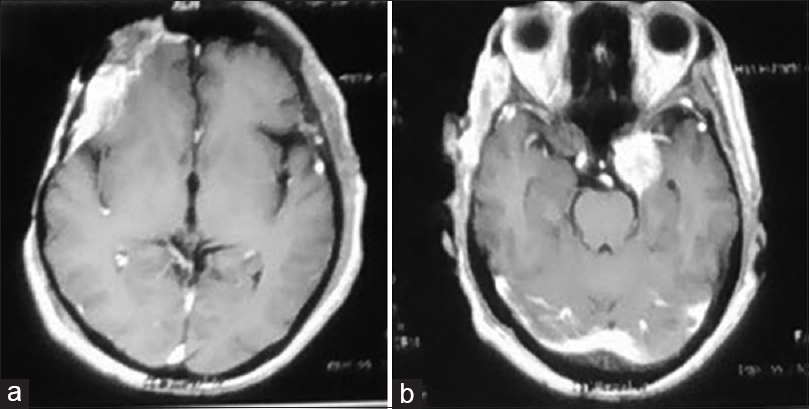
Over a period of 6 months, the patient was seen in regular follow-up when a recurrence was seen on routine imaging and also new complaint of ptosis on left side. The images show local recurrence on right frontal lobe; interestingly, a new lesion was prominent on contralateral cavernous sinus [
Case 2
A 49-year-old male patient came to our hospital with a tumorous growth located in the frontal region of his head, which developed over approximately 3 years prior to presentation. He had undergone an operation in another hospital about 2 years earlier but the lump had been growing rapidly over the preceding 6 months. Pathologic analysis revealed malignant schwannoma. On physical examination, we encountered two firm, noncompressible, nontender, nonpulsatile masses that measured approximately 6 × 5 cm and 8 × 8 cm in the frontal region with head CT scan reveal bone discontinuity due to bony destruction [
There were no clinical signs suggesting neurofibromatosis and the family history was negative. The patient underwent microsurgical resection via a frontal craniotomy followed by plastic and reconstructive in a single surgery. The involved bone and a 2-cm margin of healthy tissue were excised together with the tumor mass. The involved bone was brittle and soft, so it rongeured until thick and healthy bone was encountered. There was no infiltration of the underlying dura mater and the lesion could be completely excised en bloc. Cranioplasty was required to close the resulting 4 × 4 cm calvarial defect. After excising the mass, the scalp defect measured approximately 15 × 12 cm and the reconstruction was completed using a myocutaneous flap with a muscle cuff along with the vascular pedicle [Figure
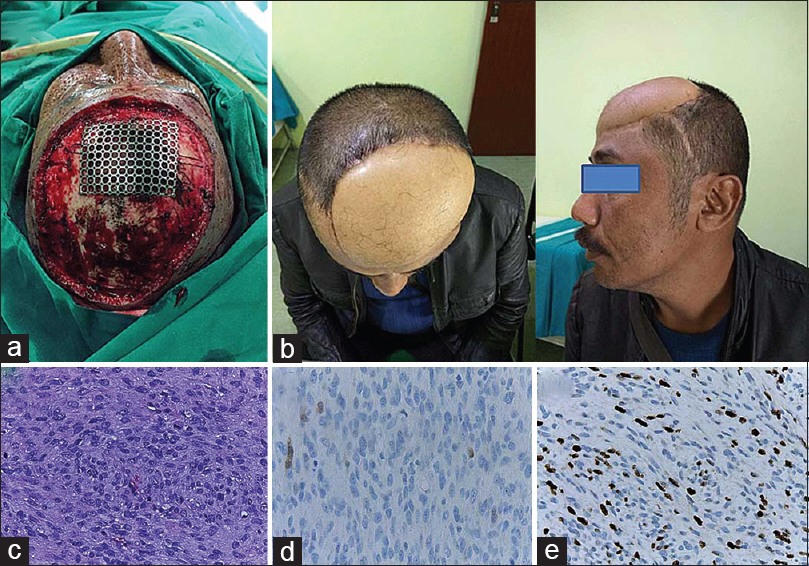
Figure 6
Intraoperative findings. (a) A cranioplasty measuring 4 × 4 cm was performed with a large scalp defect. (b) Postoperative image after 6 months. (c) High-cellular density of spindle-shaped tumor cells with frequent mitotic figures (high power fields with hematoxylin and eosin stain). (d) S-100 immuno-histochemistry, IHC. (e) Ki-67 IHC
DISCUSSION
The scalp consists of five layers: skin, connective tissue, aponeurosis (epicranial tissue), loose connective tissue, and periosteum. Any of these tissue layers may be associated with tumors, either benign or malignant.[
MPNSTs are rare tumors with an incidence of approximately 0.001% in the general population with slight male predominance and accounting for 5–10% of all soft tissue sarcomas.[
The etiology is unknown but they have a known association with NF-1,[
The initial clinical features of MPNST of the scalp are somewhat atypical when compared to other peripheral MPNSTs. Most patients present with a gradually increasing mass in the scalp (cutaneous or subcutaneous). Symptoms of headache, vomiting, seizures, vertigo, visual impairment, or focal neurological deficits may occur in patients with giant MPNST of the scalp if there is any intracranial extension of the tumor.[
Diagnostic imaging plays a significant a role in determining tumor resectability, assessing surgical risks, and in planning reconstruction. Sequential MRI is necessary for assessing the patient for possible tumor recurrence. CT scan imaging shows the extent of bony involvement, as in both the cases there was destruction of cortical bone. MRI is the investigative tool of choice because it defines the anatomic relationships between the lesion and the adjacent soft tissues, including muscular, vascular, and neural structures.[
Microscopically, MPNST is a densely cellular tumor that shows fascicular areas with alternate myxoid regions. The swirling arrangement of intermixed dense and myxoid areas has been described as a marbleized pattern.[
The current management of MPNST is comparable to that of other soft tissue tumors.[
MPNSTs are aggressive malignancies treated in the same way as other high-grade sarcomas. There are no specific guidelines for MPNST of the head or scalp. These guidelines usually follow soft tissue sarcomas. For those patients with resectable disease, a wide excision through normal uninvolved tissues is the surgical procedure of choice. Defining a rts and a literature reversial, but with the addition of effective adjuvant therapy (e.g. radiotherapy) a tumor-free margin (R0) may be adequate. Where a wide excision is not possible due to anatomical constraints, a planned marginal or microscopically positive margin against a critical structure, plus radiotherapy, for intermediate and high-grade tumors, may be an appropriate means of achieving tumor control while maintaining physical function.[
Although complete excisions can seemingly be achieved during surgery, local recurrence occurs frequently and will need further radiotherapy and/or chemotherapy. Local recurrences have been reported in as many as 52–88.9% of MPNSTs for different sites.[
CONCLUSION
We describe two rare cases of giant primary MPNST of the scalp that were treated surgically followed by radiotherapy. MPNST should be considered as a differential diagnosis of an enlarging scalp soft tissue lesion and these tumors can show bony and intracranial involvement. MPNSTs are aggressive lesion and a multimodal approach is recommended for management.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Baehring JM, Betensky RA, Batchelor TT. Malignant peripheral nerve sheath tumor: The clinical spectrum and outcome of treatment. Neurology. 2003. 61: 696-8
2. Bouhafa T, Elmazghi A, Baissel H, Fatmi HE, Amarti A, Hassouni K. Malignant peripheral nerve sheath tumors of the scalp: Case report and review of literature. Int J Clin Med. 2014. 5: 916-20
3. Dangoor A, Seddon B, Gerrand C, Grimer R, Whelan J, Judson I. UK guidelines for the management of soft tissue sarcomas. Clin Sarcoma Res. 2016. 6: 20-
4. Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986. 57: 2006-21
5. Evans DG, Huson SM, Birch JM. Malignant peripheral nerve sheath tumours in inherited disease. Clin Sarcoma Res. 2012. 2: 17-
6. Farid M, Demicco EG, Garcia R, Ahn L, Merola PR, Cioffi A, Maki RG. Malignant peripheral nerve sheath tumors. Oncologist. 2014. 19: 193-201
7. George E, Swanson PE, Wick MR. Malignant peripheral nerve sheath tumors of the skin. Am J Dermatopathol. 1989. 11: 213-21
8. Guo A, Liu A, Wei L, Song X. Malignant peripheral nerve sheath tumors: differentiation patterns and immunohistochemical features-a mini-review and our new findings. J Cancer. 2012. 3: 303-9
9. Hajdu SI. Peripheral nerve sheath tumors. Histogenesis, classification, and prognosis. Cancer. 1993. 72: 3549-52
10. Jhawar SS, Mahore A, Goel N, Goel A. Malignant peripheral nerve sheath tumour of scalp with extradural extension: Case report. Turk Neurosurg. 2012. 22: 254-6
11. Kudo M, Matsumoto M, Terao H. Malignant nerve sheath tumor of acoustic nerve. Arch Pathol Lab Med. 1983. 107: 293-7
12. Kumar P, Jaiswal S, Agrawal T, Verma A, Datta NR. Malignant peripheral nerve sheath tumor of the occipital region: Case report. Neurosurgery. 2007. 61: E1334-5
13. Latham K, Buchanan EP, Suver D, Gruss JS. Neurofibromatosis of the head and neck: Classification and surgical management. Plast Reconstr Surg. 2015. 135: 845-55
14. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella Branger D, Cavenee WK. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016. 131: 803-20
15. Teles F, Ataíde AMM, De Lima BA, Costa TCC, Lins RC, Barbosa GHTS. Giant malignant peripheral nerve sheath tumor of the scalp. Acta Neurol Taiwan. 2012. 21: 133-5
16. Wanebo JE, Malik JM, VandenBerg SR, Wanebo HJ, Driesen N, Persing JA. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 28 cases. Cancer. 1993. 71: 1247-53
17. Wang J, Ou SW, Guo ZZ, Wang YJ, Xing DG. Microsurgical management of giant malignant peripheral nerve sheath tumor of the scalp: Two case reports and a literature review. World J Surg Oncol. 2013. 11: 269-
18. Woodruff JM, Selig AM, Crowley K, Allen PW. Schwannoma (neurilemoma) with malignant transformation. A rare, distinctive peripheral nerve tumor. Am J Surg Pathol. 1994. 18: 882-95

