

- Department of Neurosurgery, King Saud Medical City, Riyadh, Saudi Arabia.
- Department of Adult Neurosurgery, Neuroscience National Institute, King Fahad Medical City, Riyadh, Saudi Arabia.
Correspondence Address:
Raghad Hany Salem, Department of Neurosurgery, King Saud Medical City, Riyadh, Saudi Arabia.
DOI:10.25259/SNI_213_2023
Copyright: © 2023 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Raghad Hany Salem1, Othman T. Almutairi2, Mohammed Saeed Bafaquh2. Multifocal malignant peripheral nerve sheath tumor in patients with neurofibromatosis type I: Report of two cases and review of literature. 28-Jul-2023;14:261
How to cite this URL: Raghad Hany Salem1, Othman T. Almutairi2, Mohammed Saeed Bafaquh2. Multifocal malignant peripheral nerve sheath tumor in patients with neurofibromatosis type I: Report of two cases and review of literature. 28-Jul-2023;14:261. Available from: https://surgicalneurologyint.com/surgicalint-articles/12469/
Date of Submission
05-Mar-2023
Date of Acceptance
14-May-2023
Date of Web Publication
28-Jul-2023
Abstract
Background: Malignant peripheral nerve sheath tumors (MPNSTs) are one of the rarest soft-tissue sarcomas with a prevalence of 0.001% in the general population. It is closely associated with a unique neurocutaneous stigmata under the spectrum of the dermatological manifestations of neurofibromatosis type 1 (NF1). Almost 81% of MPNST arises from a precursor neuroma, and multifocality of these lesions is extremely rare, making up to 0.001% of cases. Moreover, spinal cases are extremely uncommon with only four cases reported internationally. Here, we present the fifth and sixth spinal MPNST cases with a brief review of literature.
Case Description: We describe two unusual cases of multifocal MPNST in relation to NF1 occurring in the spinal cord. Both patients presented with local pain and myelopathic symptoms. The two patients underwent wide surgical resection, followed by neoadjuvant radiotherapy and reported immediate postoperative improvement of the presented complaint; however, one patient suffered from rapid recurrence and metastasis.
Conclusion: Due to the scarcity of spinal cases related to MPNST, no clear guidelines regarding the management of these cases are set in the literature. Histopathological diagnosis remains as the most pivotal diagnostic tool as they can mimic other peripheral nerve sheath lesions, such as neuromas and schwannomas, in imaging. Cases that were managed by early surgical intervention in addition to neoadjuvant radiotherapy reported the best outcome. However, cases of MPNST in concomitance with NF1 were found to be resistant to both chemo and radiotherapy and have high recurrence rate.
Keywords: Malignant peripheral nerve sheath tumor, Neurofibromatosis, Neuro-oncology, Neurosarcoma, Peripheral nerve tumor
INTRODUCTION
Neurofibromatosis is a neurocutaneous syndrome that causes the individual to develop a wide spectrum of pathologies, including multiple painless nodules, referred to as neurofibromas, as a result of mutation – both as inherited autosomal dominant trait or sporadic mutation – in the tumor-suppressor gene. There are two types of this syndrome: Type 1 and 2. Neurofibromatosis type 1 (NF1), also known as Recklinghausen syndrome, is the most prevalent of the two with an incidence rate of 1:2,500–3,000.[
CASE DESCRIPTION
Case 1
A 50-year-old male who was medically and surgically free with cutaneous stigmata of neurofibromatosis type I since birth. The patient presented with 3 months’ history of backache and left femoral nerve distribution neuropathic pain and claudication with intact sensory/motor/sphincter performance. On examination, he was found to have cutaneous stigmata of NF1, congenital left 6th cranial nerve palsy, and left posterior thigh mass; but otherwise, he was neurologically intact with no long tract signs. A radiological assessment with magnetic resonance imaging (MRI) brain scan showed diffusely increased leptomeningeal enhancement and multiple numerous enhancing nodules in the scalp. An MRI of the cervical spine displayed a hyperintense well-circumscribed mass in the right carotid space in the neck extending from C1 to T1 levels. It measures 4.8 × 4.9 cm in the anterior-posterior and transverse planes, respectively [
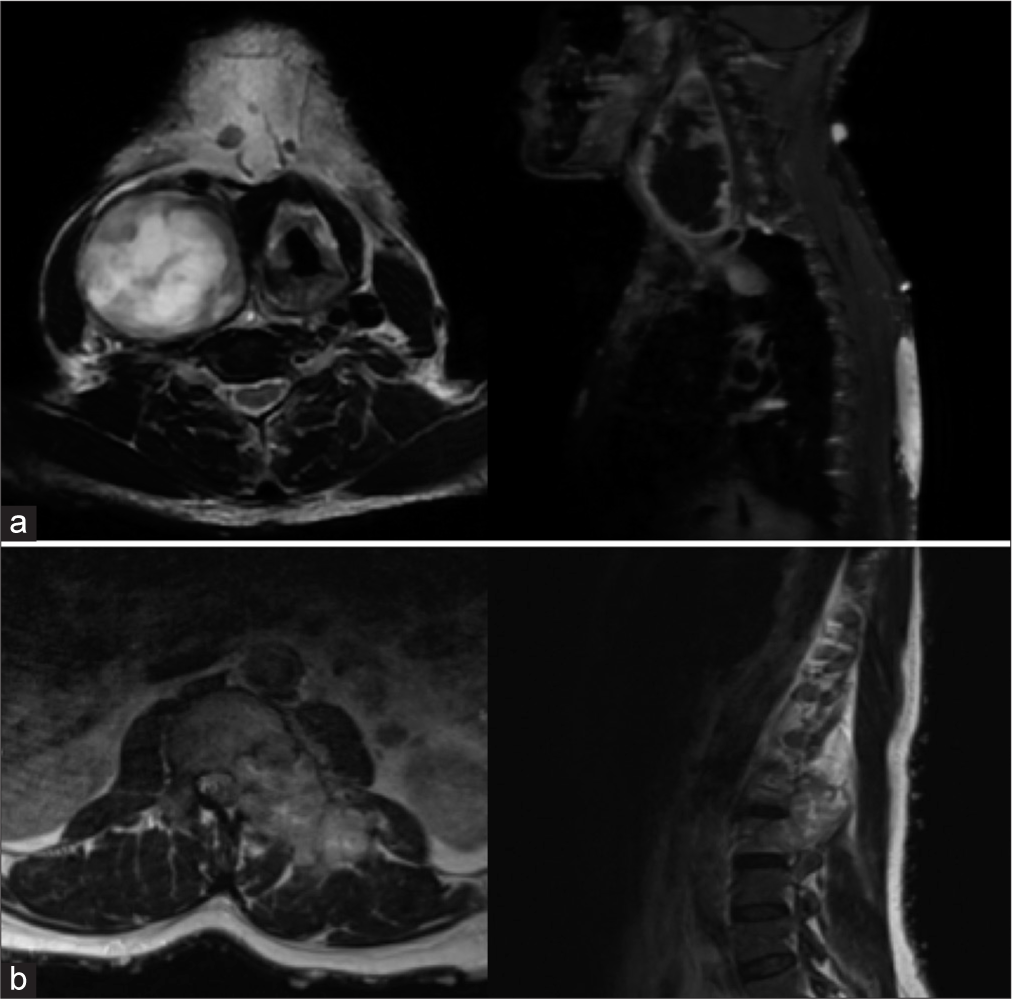
Figure 1:
(a) Preoperative axial and sagittal cut of magnetic resonance imaging (MRI) (T2) cervical spine showing a well-defined heterogeneous mass in the right carotid space in the neck extending from C1 to T1 levels. (b) Preoperative axial and sagittal cut of MRI (T2) lumbar spine showing another a large intraspinal extradural mass with similar intensity in the lumbar spine at the level of L1–L3. There is scalloping and sclerosis as well as abnormal enhancement of the L2 vertebral body secondary to tumor involvement.
Case 2
Patient information
A 27-year-old female who was recorded to have NF1 syndrome 6 years ago, who underwent two previous surgeries for the removal of peripheral neural tumor grade II in the left arm and the right retropharyngeal space, and is currently being followed for spinal neuroma, presented with an ongoing right lower limb weakness for 7 months now. An examination of her appearance revealed a healthy-looking female with multiple cafe au lait spots. Examination of the left lower limb revealed power of -4/5 all over and hyporeflexia. The rest of the neurological examination was unremarkable. Her abdomen was soft and lax with a palpable solid mass in the right upper quadrant extending to the right flank area with minimal tenderness. An MRI brain scan showed a stable hyperintensity focus on T2 within the left occipital lobe, which was stable since November 2016 and a bilateral globus pallidus hamartoma that is larger in the right side [
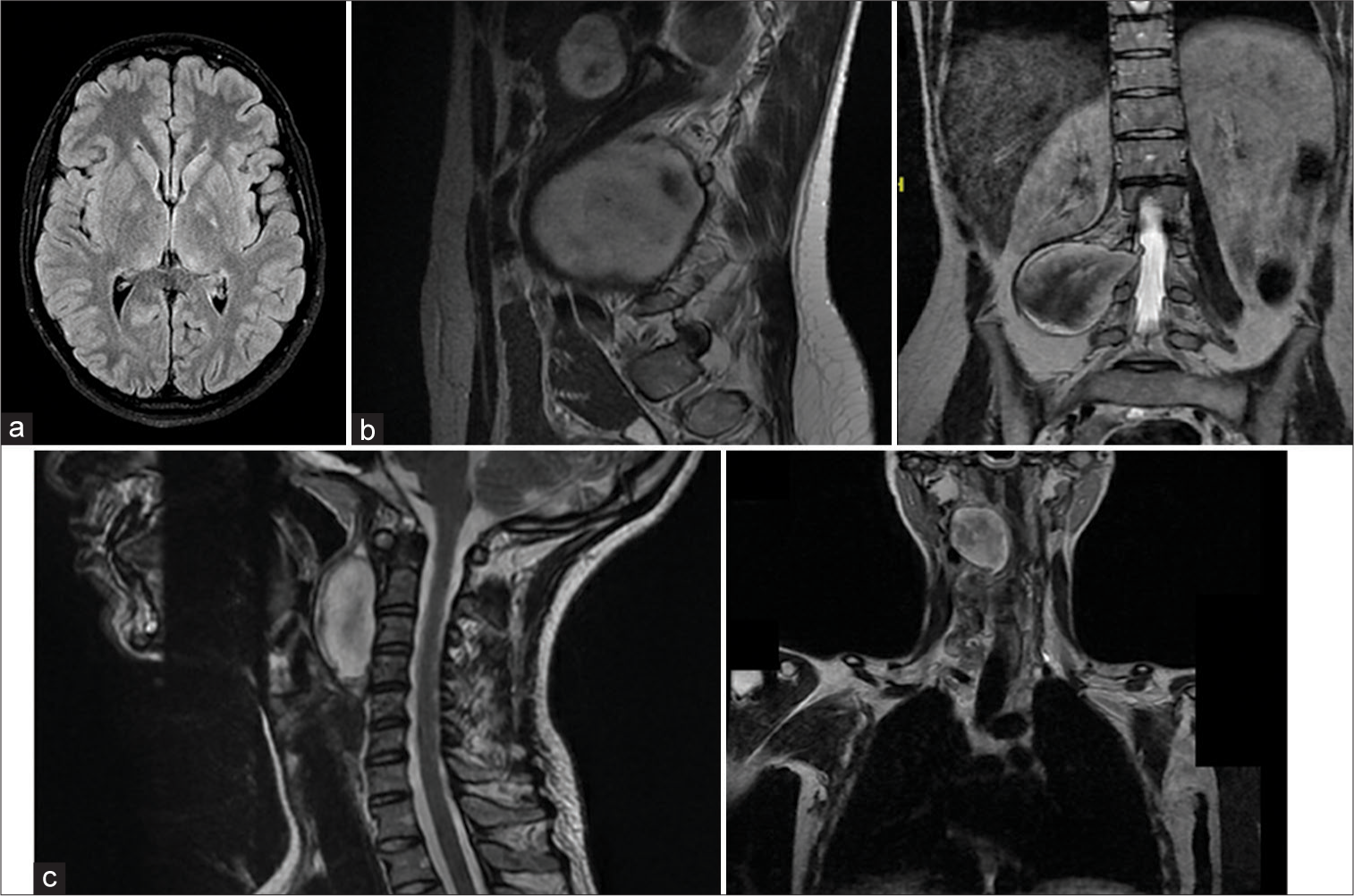
Figure 2:
(a) Axial cut of magnetic resonance imaging (MRI) brain (fluid-attenuated inversion recovery) showing bilateral hamartoma in globus pallidus. (b) Preoperative sagittal and coronal cut MRI T2 of lumbar spine showing an ill-defined mass at the level of L3–5. (c) Preoperative sagittal and coronal cut MRI T2 of the cervical spine showing a predominantly hypodense well-defined mass found in the right retropharyngeal space.
DISCUSSION
MPNSTs, previously known as neurological sarcoma or neurofibrosarcoma, are rare, aggressive, and rapidly progressive soft-tissue carcinoma with an incident of only 0.001% in the general population.[
Clinical presentation varies depending on the location and size of the MPNST. Most commonly, patients will present with rapidly expanding mass, neuropathic pain, and local neurological defects (weakness or paresthesia). The duration of the onset of symptoms ranges from months to years[
Establishing an accurate diagnosis from imaging is a major challenge. Contrasted MRI is the preferred modality in diagnosing and surgical planning of MPNST. Some case reports even utilized whole body MRI (WB-MRI) to identify any synchronous lesions for grading.[
As for the diagnostic workup, there are many techniques in retaining a tissue biopsy with no specific guidelines; however, excisional biopsy showed the least amount of false negative as other modalities may only show segments of the precursor benign lesion.[
Early wide surgical resection remains pivotal in the treatment of MPNST. Cases that were managed with subtotal resection combined with adjuvant radiotherapy resulted in rapid recurrence and metastasis and ended with mortality. Wide excision alone achieved 43% survival rate; on the other hand, combining it with high-dose radiation achieved the best outcome with the lowest recurrence rates for patients. Thus, the latest literature suggests that adjuvant radiation should be given to all MPNST regardless of the stage of the disease to reduce the recurrence rates. It is important to note that some cases that were treated with local radiation later on developed radiation-induced sarcomas; as such, close follow-up is of extreme importance. For peripheral MPNST, amputation is considered the definitive management, but most cases ended in mortality as surgeries were delayed.[
MPNST generally carry poor prognosis, with high mortality rates reaching up to 68%[
CONCLUSION
MPNST is a rare tumor that is closely related to NF1 syndrome. It presents as a large expansile mass that causes pain and local neurological defects due to its mass effect. Due to the scarcity of spinal cases involving MPNST, there is insufficient clinical experience that can be based on to set guidelines in their management. Moreover, WB-MRI proved to be the most useful modality in both diagnosis and grading. To this day, the gold standard in diagnosis remains to be histopathology. Radical excision with wide safe margins followed by adjuvant radiotherapy has the highest success rates in the reported cases. Further investigation is required to help better understand the behaviors of such obscure lesions and the future means of its management.
Declaration of patient consent
Patients’ consent not required as patients’ identities were not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
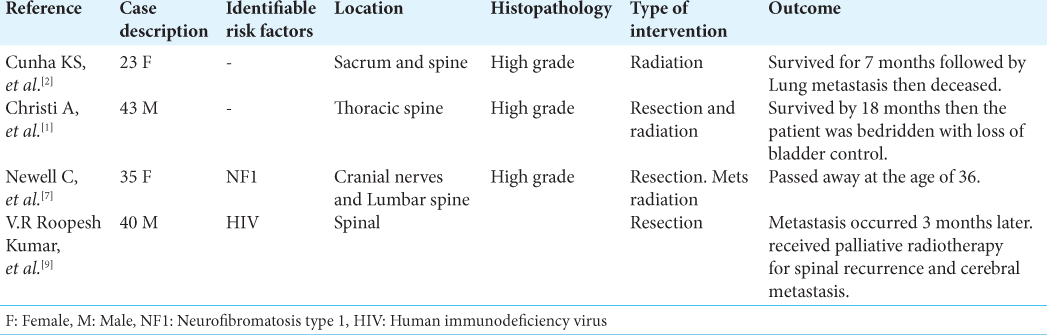
1. Christi AY, Baskoro W, Kameswari B, Hakim IA, Pangaribuan VS, Purnawan A. Extradural malignant peripheral nerve sheath tumor of the thoracic spine: A rare case report. Surg Neurol Int. 2021. 12: 560
2. Cunha KS, Caruso AC, Faria PA, Silva LE, Pires AR, Geller M. Malignant peripheral nerve sheath tumors: Clinicopathological aspects, expression of p53 and survival. Clinics (Sao Paulo). 2012. 67: 963-8
3. Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986. 57: 2006-21
4. Farid M, Demicco EG, Garcia R, Ahn L, Merola PR, Cioffi A. Malignant peripheral nerve sheath tumors. Oncologist. 2014. 19: 193-201
5. Higami Y, Shimokawa I, Kishikawa M, Okimoto T, Ohtani H, Tomita M. Malignant peripheral nerve sheath tumors developing multifocally in the central nervous system in a patient with neurofibromatosis Type 2. Clin Neuropathol. 1998. 17: 115-20
6. Kroep JR, Ouali M, Gelderblom H, Le Cesne A, Dekker T, Van Glabbeke M. First-line chemotherapy for malignant peripheral nerve sheath tumor (MPNST) versus other histological soft tissue sarcoma subtypes and as a prognostic factor for MPNST: An EORTC soft tissue and bone sarcoma group study. Ann Oncol. 2011. 22: 207-14
7. Kumar VR, Madhugiri VS, Sasidharan GM, Ganesh CV, Gundamaneni SK. Multifocal spinal malignant peripheral nerve sheath tumor in an immunocompromised individual: Case report and review of literature. Eur Spine J. 2014. 23: 236-41
8. Newell C, Chalil A, Langdon KD, Karapetyan V, Hebb MO, Siddiqi F. Cranial nerve and intramedullary spinal malignant peripheral nerve sheath tumor associated with neurofibromatosis-1. Surg Neurol Int. 2021. 12: 630
9. Porter DE, Prasad V, Foster L, Dall GF, Birch R, Grimer RJ. Survival in malignant peripheral nerve sheath tumours: A comparison between sporadic and neurofibromatosis Type 1-associated tumours. Sarcoma. 2009. 2009: 756395
10. Scheithauer BW, Erdogan S, Rodriguez FJ, Burger PC, Woodruff JM, Kros JM. Malignant peripheral nerve sheath tumors of cranial nerves and intracranial contents: A clinicopathologic study of 17 cases. Am J Surg Pathol. 2009. 33: 325-38
11. Shofty B, Barzilai O, Khashan M, Lidar Z, Constantini S. Spinal manifestations of Neurofibromatosis Type 1. Childs Nerv Syst. 2020. 36: 2401-8
12. Steins MB, Serve H, Zuhlsdorf M, Senninger N, Semik M, Berdel WE. Carboplatin/etoposide induces remission of metastasised malignant peripheral nerve tumours (malignant schwannoma) refractory to first-line therapy. Oncol Rep. 2002. 9: 627-30

