

- Department of Neurosciences, Institute of Translational Neurosciences, CUCS University of Guadalajara, Jalisco, México
- Department of Neurosciences, CUCS University of Guadalajara, Jalisco, México
Correspondence Address:
Rodrigo Ramos-Zúñiga
Department of Neurosciences, CUCS University of Guadalajara, Jalisco, México
DOI:10.4103/2152-7806.166889
Copyright: © 2015 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Rodrigo Ramos-Zúñiga, Daniel Alexander Saldaña-Koppel. Neurofibromatosis type 1 and pregnancy: The transformation of a nodular to cystic neurofibroma in the cervical region. Surg Neurol Int 08-Oct-2015;6:
How to cite this URL: Rodrigo Ramos-Zúñiga, Daniel Alexander Saldaña-Koppel. Neurofibromatosis type 1 and pregnancy: The transformation of a nodular to cystic neurofibroma in the cervical region. Surg Neurol Int 08-Oct-2015;6:. Available from: http://surgicalneurologyint.com/surgicalint_articles/neurofibromatosis-type-1-and-pregnancy-the-transformation-of-a/
Abstract
Background:The peripheral hallmarks of neurofibromatosis type 1 (NF1) are Café au lait and solid nodular neurofibromas. The morphological behavior of these lesions could be susceptible to modification during pregnancy. The present case report describes a case of cystic transformation of a nodular neurofibroma, with progressive growth and mass effect in the anterior cervical region, which was surgically resolved without any complications.
Case Description:A 33-year-old female patient with a known personal history of NF1, with annual control of the peripheral neurofibromas and cerebral and spinal magnetic resonance imaging follow-ups. Under genetic counseling, she decides to get pregnant following all the medical advises. Once the pregnancy is confirmed, she starts to notice the growth of one of them adjacent to the left cervical region. Such neurofibroma presented with the progressive gradual increase and in the last month, she presented dysphagia, dysphonia, and postural pain localized by the mass effect. Once the pregnancy concluded, the microsurgical approach was scheduled for resection of the lesion, where a cystic mass was found within the walls of the neurofibroma. The resection was uneventful.
Conclusion:The transformation of a nodular to cystic neurofibroma during pregnancy is a very rare presentation, which may exacerbate the clinical symptomatology depending on the topography of the lesion due to the mass effect it may create. This condition may alert to the recommendations and vigilance in patients with NF1, who are pregnant or are planning on a future pregnancy. The neurosurgical resolution in this region is safe and beneficial.
Keywords: Microsurgery, neck dissection, neurofibromatosis type 1, neuropathology, pregnancy
INTRODUCTION
Neurofibromatosis type 1 (NF1) is one of the genetic neurocutaneous syndromes that has a greater number of phenotypic expressions.[
The clinical criteria for the disease are determined by the presence of six or more café-au-lait macules, >5 mm in prepubertal patients and >15 mm in postpubertal patients, two or more neurofibromas of any type, at least one plexifrom neurofibroma, optical gliomas, two or more Lisch nodules, sphenoid wing dysplasia, and pseudoarthrosis.[
The NF1 gene encodes neurofibromin, a tumoral suppressor protein.[
CASE REPORT
The patient is a 33-year-old female patient with personal history of NF1 (without any family history in the two previous generations) characterized by multiple Café au lait macules and multiple neurofibroma nodules in distinct regions of the skin which involve the scalp, neck, back, abdomen, and all the extremities.
The patient has a normal evolution of her disease with no incidents; she is an independent professional and refers no other symptomatology. Eventually, she complains of pain due to the mechanical compression of one of the neurofibromas. After considering the genetic counseling, the patient decides to get pregnant. During the second trimester of her pregnancy, she started to note gradual and progressive growth of one of the neurofibromas located in the anterolateral left portion of the neck, in the angle formed by the thyroid gland and the common carotid artery. Such growth gradually increased to the point where by the end of the pregnancy it had a diameter of approximately 10 cm × 15 cm, it made swallowing difficult, dysphonia, and generated local pain (nonneuropathic pain) [
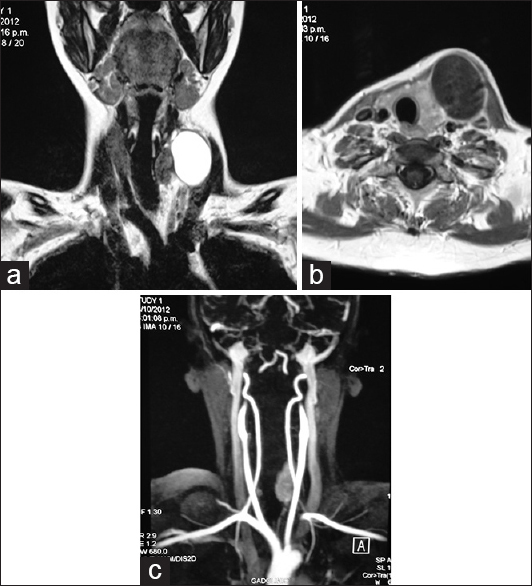
Figure 1
Magnetic resonance imaging and angiomagnetic resonance imaging. (a and b) Magnetic resonance imaging shows a cystic tumor in the left anterior cervical region creating mas effect over the midline structures. (c) There is no evidence of intramural vascular flow in the angiomagnetic resonance imaging
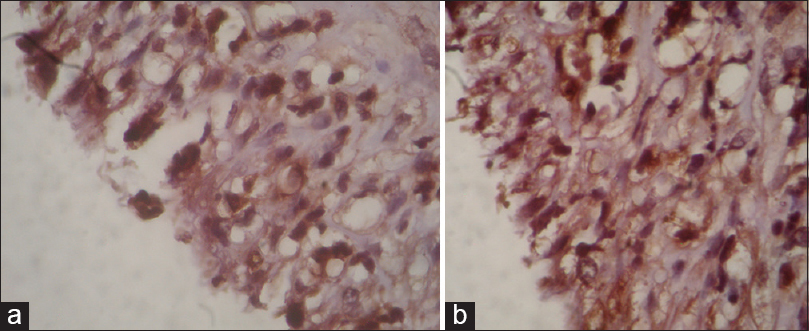
After neuroimaging evaluation, a surgical approach is decided 3 months after the C-section using general anesthesia and microsurgical dissection. A tumor mass was identified, with a superficial wall, free from vascular or cervical major nerve structures, with a clear serous liquid content that after decompression, modifies the tumoral morphology immediately, allowing identification of the layers of the cystic lesion. The visceral portion of the capsule was found attached to the external plane of the thyroid gland and to the carotid artery adventitia, which was preserved. The postsurgical evolution was normal, without any complications. There were no alterations regarding phonation or deglutition, and there was a normal recovery of the external anatomy of the neck without any evidence of tumoral mass. The analysis of the fluid reported no cytological alterations and culture was negative for infection. The hematoxylin and eosin stain shows the presence of neoplastic cells, nuclear and diffuse cytoplasmic positivity to S-100 protein [
DISCUSSION
The interaction of neurofibromin with other membrane cellular proteins such as proteoglycans, intermediate actin filaments, and tubulin;[
A recent theory that aims to validate the determined changes by the second mutations, which has been described in hormonal events such as puberty and pregnancy, has been described through the demonstration of a multipotential progenitor NF1 +/− cell, which is capable of in vitro differentiation into the different cells types found in neurofibromas, including schwann cells, fibroblasts and epithelial cells.[
This condition partially explains one of the fundamental reasons for why, in the study case, pregnancy, and the own trophic factors were determining variables in the volume growth of the neurofibroma starting on the first trimester and it also reassures the tendency of the growth heterogeneity not derived directly from the proliferation of schwann cells to express as a solid tumor, but from other cellular linages such as mast cells, fibroblasts, adipocytes, and epithelial cells which generate a liquid material, that was finally responsible for the volumetric expansion of the tumor creating mass effect.
The growth and mass effect of a neurofibroma during puberty or pregnancy is a condition that should be monitored due to the possibility of the appearance of new symptoms in patients diagnosed with NF1.[
The transformation of a cystic neurofibroma is rarely described in the literature; hence, this case represents minor complexity in the surgical approach. The planning and surgical strategy is similar to the carotid glomus surgical approach. The preservation of the adjacent vascular and neural structures is mandatory.
CONCLUSION
The stable patients diagnosed with NF1, can experiment some changes due to epigenetic factors that may second the mutation and modify the phenotypic expression. Such is the case in puberty and pregnancy due to the presence of trophic hormonal factors that may activate the cellular linage differentiation in the neurofibroma and contribute to its growth, whether it's solid or cystic. The surgical approach evaluating the risk/benefit is the most appropriate conduct for the resolution of the mass effect.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Bollag G, McCormick F, Clark R.editors. Characterization of full-length neurofibromin: Tubulin inhibits Ras GAP activity. EMBO J. 1993. 12: 1923-7
2. Bonneau F, D’Angelo I, Welti S, Stier G, Ylänne J, Scheffzek K. Expression, purification and preliminary crystallographic characterization of a novel segment from the neurofibromatosis type 1 protein. Acta Crystallogr D Biol Crystallogr. 2004. 60: 2364-7
3. Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002. 39: 311-4
4. Huson SM, Compston DA, Clark P, Harper PS. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet. 1989. 26: 704-11
5. Jadayel D, Fain P, Upadhyaya M, Ponder MA, Huson SM, Carey J. Paternal origin of new mutations in von Recklinghausen neurofibromatosis. Nature. 1990. 343: 558-9
6. Jouhilahti EM, Peltonen S, Callens T, Jokinen E, Heape AM, Messiaen L. The development of cutaneous neurofibromas. Am J Pathol. 2011. 178: 500-5
7. Jouhilahti EM, Peltonen S, Heape AM, Peltonen J. The pathoetiology of neurofibromatosis 1. Am J Pathol. 2011. 178: 1932-9
8. Lammert M, Friedman JM, Kluwe L, Mautner VF. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol. 2005. 141: 71-4
9. Legius E, Marchuk DA, Collins FS, Glover TW. Somatic deletion of the neurofibromatosis type 1 gene in a neurofibrosarcoma supports a tumour suppressor gene hypothesis. Nat Genet. 1993. 3: 122-6
10. Maertens O, Brems H, Vandesompele J, De Raedt T, Heyns I, Rosenbaum T. Comprehensive NF1 screening on cultured Schwann cells from neurofibromas. Hum Mutat. 2006. 27: 1030-40
11. Shen MH, Harper PS, Upadhyaya M. Molecular genetics of neurofibromatosis type 1 (NF1). J Med Genet. 1996. 33: 2-17
12. Stumpf D, Alksne J, Annegers J, Brown S, Conneally P, Housman D. Neurofibromatosis.Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988. 45: 575-8
13. Terry AR, Barker FG, Leffert L, Bateman BT, Souter I, Plotkin SR. Neurofibromatosis type 1 and pregnancy complications: A population-based study. Am J Obstet Gynecol. 2013. 209: 46.e1-8
14. Tidyman WE, Rauen KA. The RASopathies: Developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009. 19: 230-6
15. Upadhyaya M, Ruggieri M, Maynard J, Osborn M, Hartog C, Mudd S. Gross deletions of the neurofibromatosis type 1 (NF1) gene are predominantly of maternal origin and commonly associated with a learning disability, dysmorphic features and developmental delay. Hum Genet. 1998. 102: 591-7

