

- School of Medicine, Universidad Peruana Cayetano Heredia, Lima, Peru,
- School of Medicine, Universidad Nacional Mayor de San Marcos, Lima, Peru,
- Department of Pediatric Endocrinology, Hospital Nacional Edgardo Rebagliati Martins, Lima, Peru
- Department of Pediatric Neurosurgery, Hospital Nacional Edgardo Rebagliati Martins, Lima, Peru.
Correspondence Address:
Nicole M. Castillo-Huerta, School of Medicine, Universidad Peruana Cayetano Heredia, Lima, Peru.
DOI:10.25259/SNI_64_2023
Copyright: © 2023 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Nicole M. Castillo-Huerta1, Joanna I. Carassa de la Cruz1, Luz Quispe-Garate2, María A. Lévano-Martínez1, Bianca Miranda Cabrera3, Erick Custodio Sheen4. Neurosurgical aspects and clinical outcomes on the treatment of Cushing disease in pediatric patients: Case series and literature review. 07-Apr-2023;14:123
How to cite this URL: Nicole M. Castillo-Huerta1, Joanna I. Carassa de la Cruz1, Luz Quispe-Garate2, María A. Lévano-Martínez1, Bianca Miranda Cabrera3, Erick Custodio Sheen4. Neurosurgical aspects and clinical outcomes on the treatment of Cushing disease in pediatric patients: Case series and literature review. 07-Apr-2023;14:123. Available from: https://surgicalneurologyint.com/?post_type=surgicalint_articles&p=12246
Date of Submission
19-Jan-2023
Date of Acceptance
16-Mar-2023
Date of Web Publication
07-Apr-2023
Abstract
Background: Cushing disease (CD) is a state of hypercortisolism caused by an adrenocorticotropic hormone-(ACTH) producing pituitary adenoma which rarely occurs in pediatric patients. The outstanding features are weight gain and growth retardation. However, the insidious onset and rarity of the disease in children and adolescents often result in delayed diagnosis.
Case Description: We present five patients
Conclusion: CD is a heterogeneous disorder that requires multidisciplinary diagnosis and management. Transsphenoidal selective adenomectomy is the optimal treatment because of its higher remission rates. However, technical and anatomic aspects should be considered in pediatric patients.
Keywords: Pediatric, Pituitary adrenocorticotropic hormone hypersecretion, Transcranial surgery, Transsphenoidal surgery, Treatment outcome
INTRODUCTION
Cushing syndrome is a hormonal disorder due to prolonged cortisol exposure. The most common cause in the pediatric population, as at any age, is exogenous administration of glucocorticoids. On the other hand, endogenous hypercortisolism is a rare condition in children, but when it occurs, an ACTH-secreting pituitary adenoma, known as Cushing disease, is the most common finding.[
RESULTS
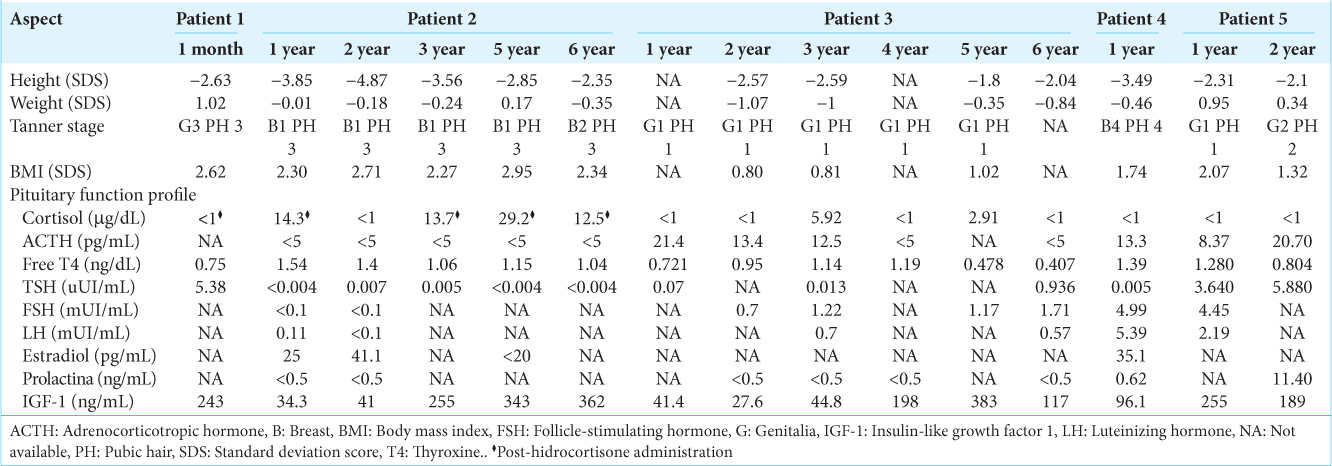
Five patients aged <14 years underwent neurosurgery for CD in the Department of Pediatric Neurosurgery at the “Hospital Nacional Edgardo Rebagliati Martins” in Lima, Peru, between 2016 and 2022. Age at diagnosis ranged from 5.5 to 12.5 years with a history of disease from 9 months to 3.5 years of moderate to severe stunting and obesity, among other features associated with CS, such as cushingoid facies, cervical hump, and acanthosis nigricans [
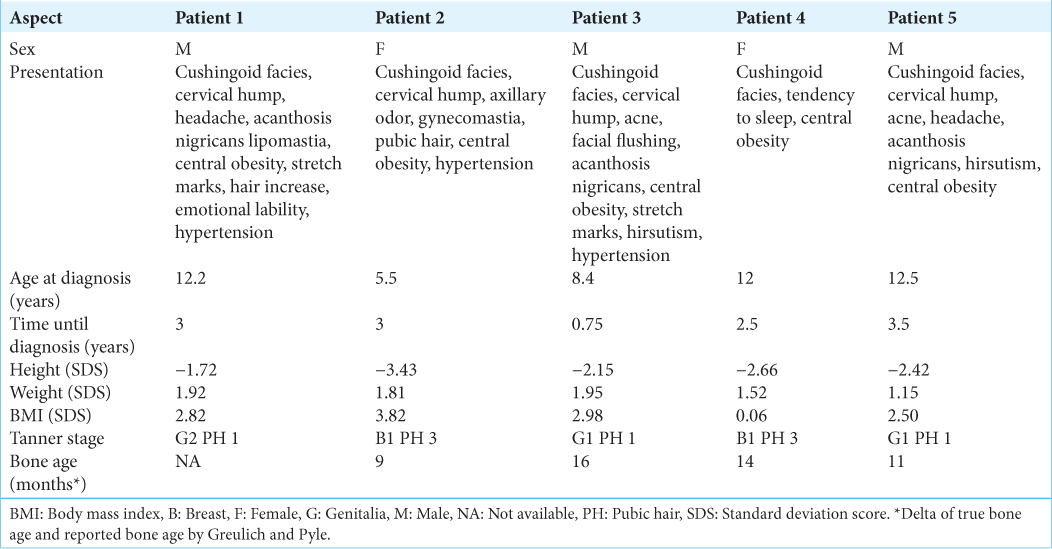
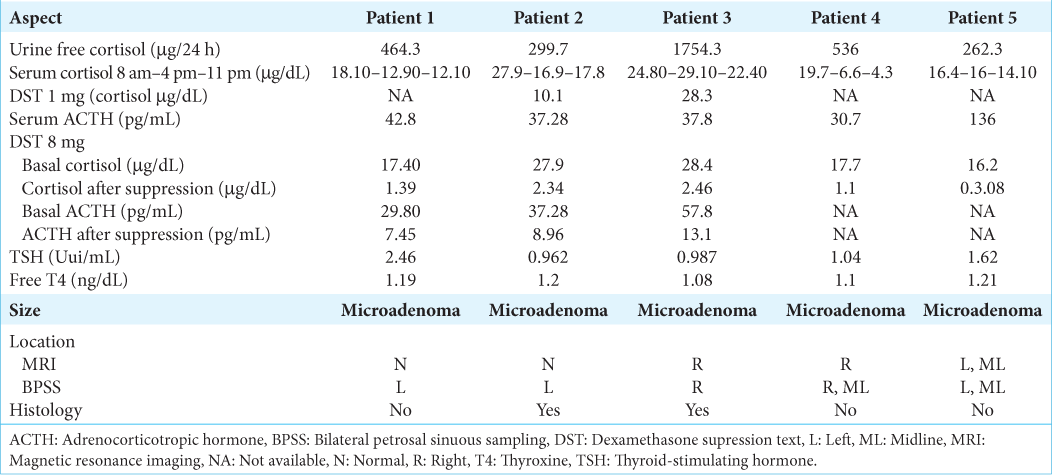
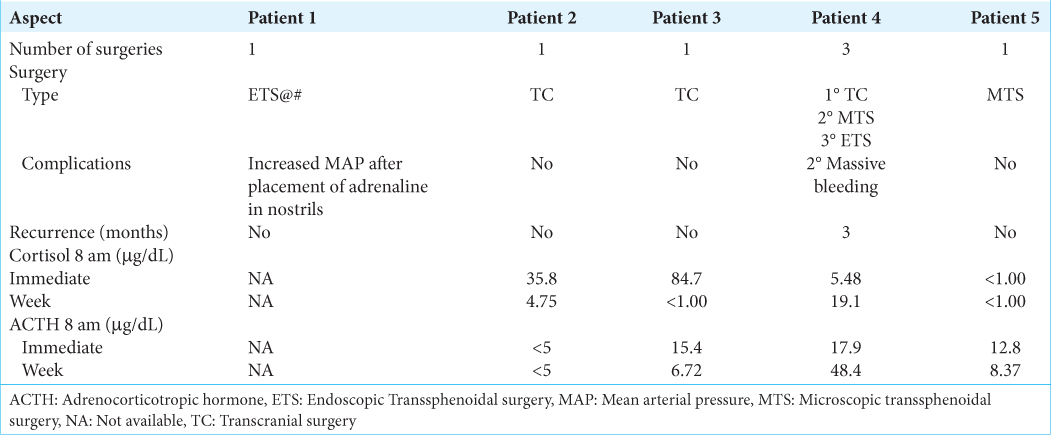
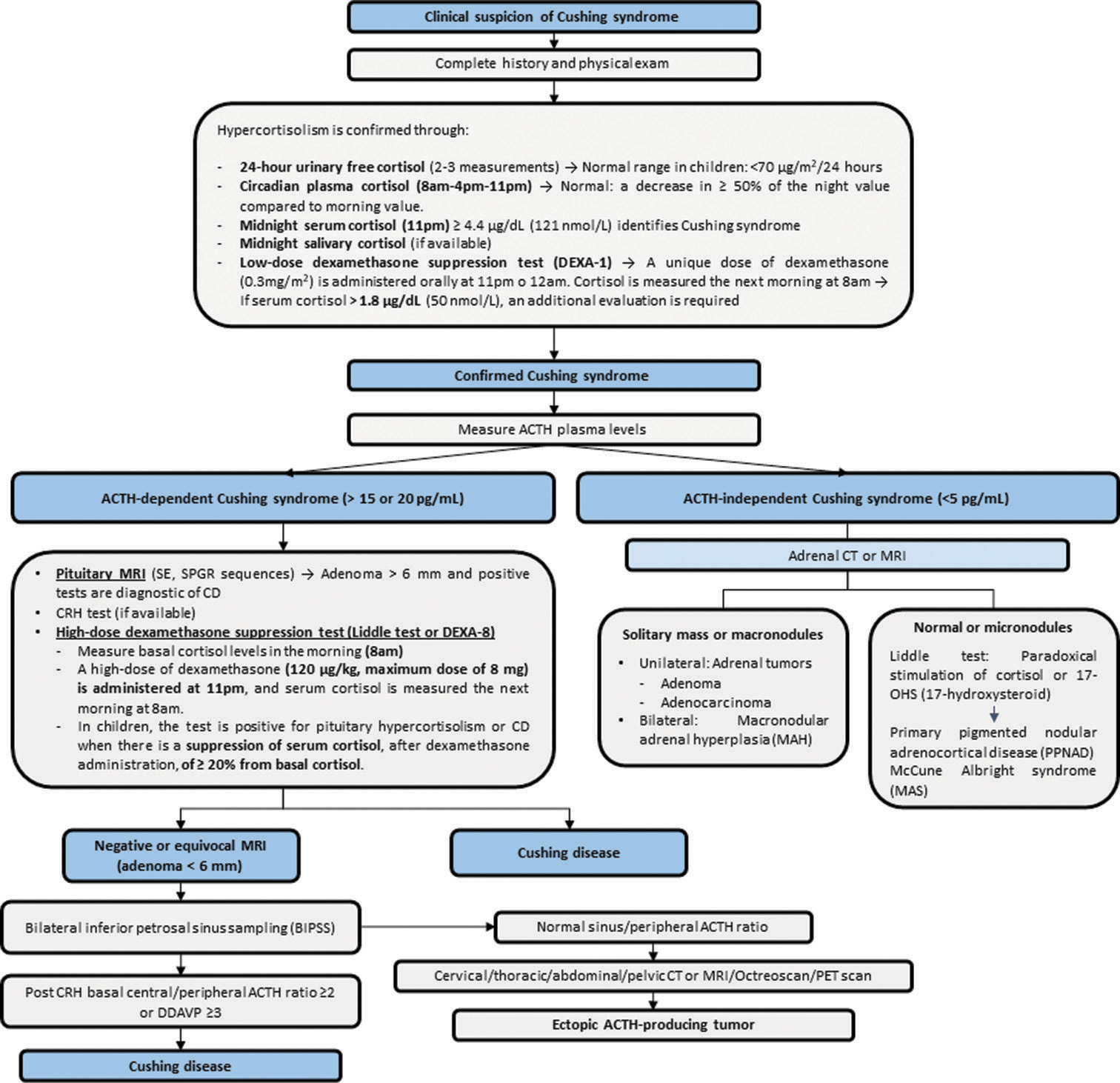
Figure 1:
Diagnostic algorithm for Cushing disease in pediatric patients. ACTH: Adrenocorticotropic hormone, CD: Cushing disease, CRH: Corticotropin-releasing hormone, CT: Computed tomography, DDAVP: desmopressin, MRI: Magnetic resonance imaging, PET: Positron emission tomography, SE: Spin echo, SPGR: Spoiled gradient-recalled acquisition.
CASE 1
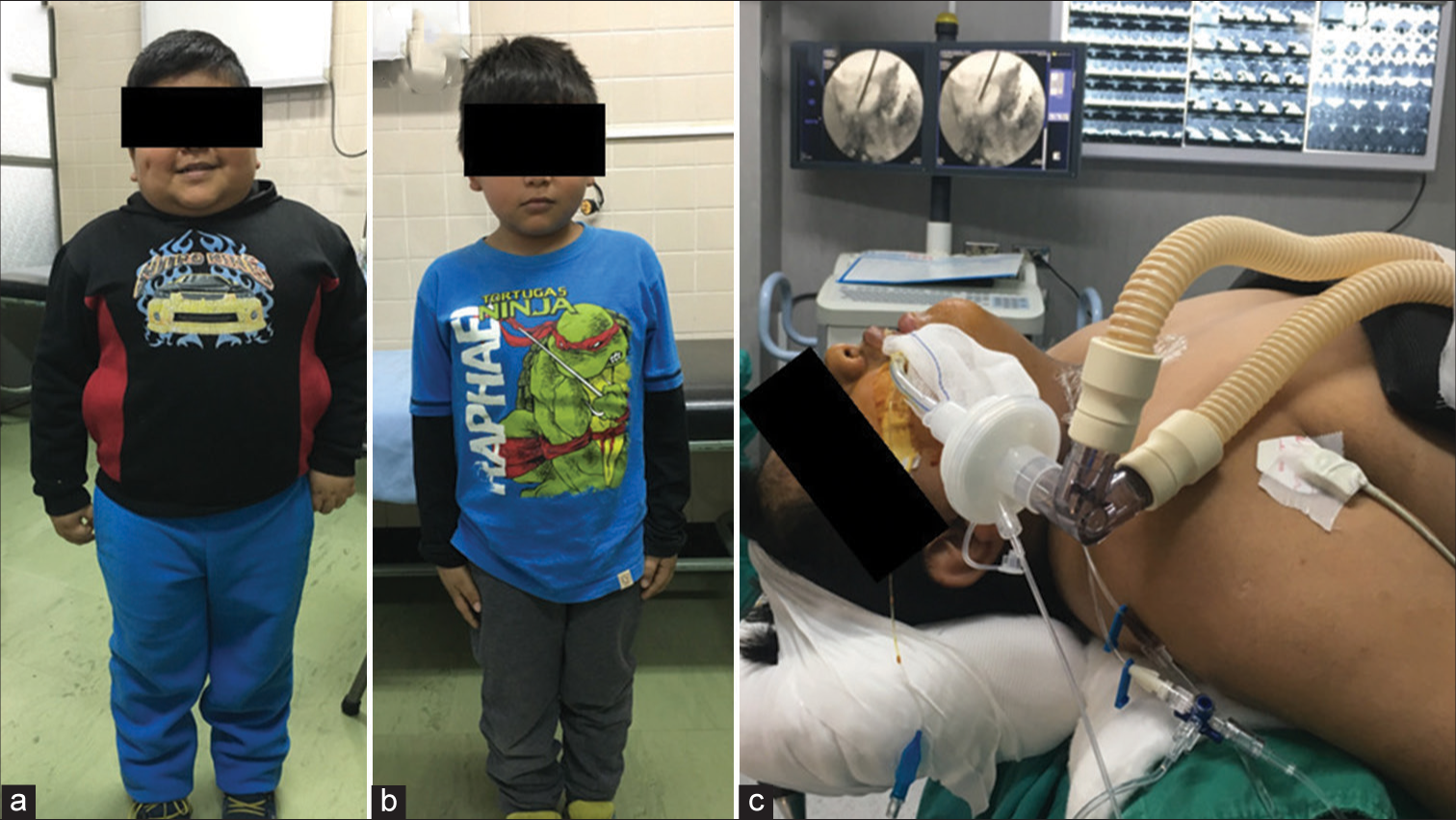
A 13-year-old male child presented to the outpatient consultation because he had been ill for 3 years. His mother reported weight gain, darkening of the posterior neck, the appearance of a cervical hump, and episodic headaches that were later accompanied by tinnitus. His clinical presentation consisted of a cushingoid facies, hair proliferation, acanthosis nigricans, a cervical hump, lipomastia, central obesity, headache, tinnitus, abdominal stretch marks, and hypertension. Serologic testing confirmed endogenous hypercortisolism, although magnetic resonance imaging (MRI) was found to be normal. Subsequently, the petrosal sinus sampling revealed a right-sided pituitary adenoma. Two years after diagnosis, a complete resection of a microadenoma through an endoscopic transsphenoidal approach. Apart from an increase in mean arterial blood pressure, which was satisfactorily treated, no complications occurred. The patient progressed favorably and is currently under endocrinological monitoring. Since surgery was recently performed, follow-up after 1 month is offered [
CASE 2
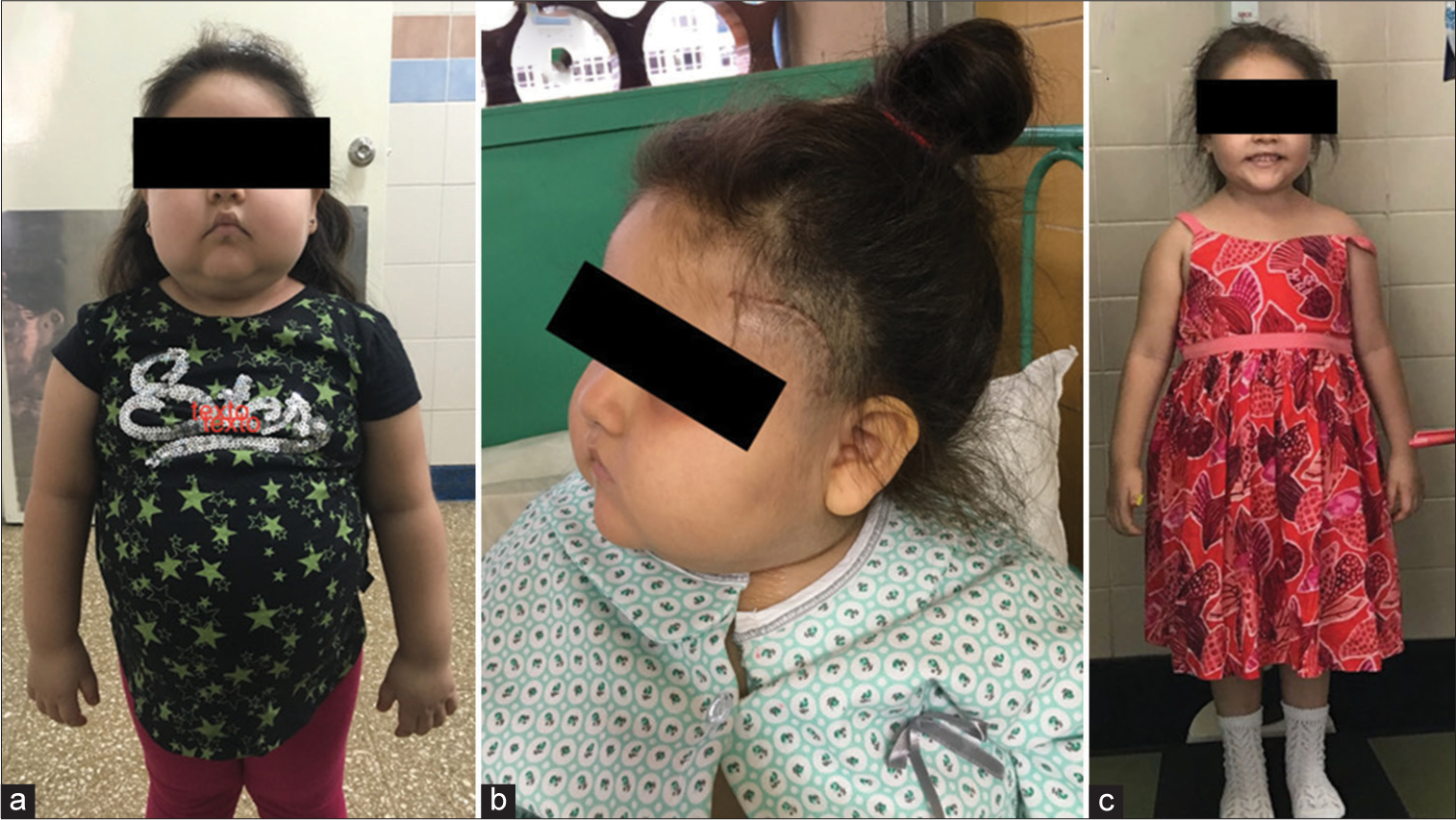
A 5-year-old female child presented to the outpatient clinic because she had been ill for 3 years. The mother reported having noticing body and facial changes, and axillary odor. On physical examination, the child presented cushingoid facies, facial flushing, a cervical hump, gynecomastia, pubic hair, central obesity, short stature, and hypertension. In suspicion of hypercortisolism, the complete blood test algorithm was held and CD was diagnosed. The MRI was interpreted as normal. The diagnosis was definitively confirmed through the petrosal sinus sampling, which demonstrated a left-sided pituitary corticotroph adenoma. One month after diagnosis, a complete excision of the pituitary gland was performed via a transcranial approach (mini-pterional craniotomy). The patient did not have a recurrence, and she continues her follow-up visitis with the pediatric endocrinologist for postsurgical hypopituitarism. After 2 years, due to the patient’s persistent short stature, growth hormone therapy was initiated.
CASE 3
An 8-year-old male child presented to the outpatient consultation with a 2-year history of illness. The mother reported weight gain, hair growth, facial acne, and axillary odor. On physical examination, the child showed central obesity, cushingoid facies, a cervical hump, facial flushing, acne, acanthosis nigricans, hirsutism, abdominal stretch marks, and hypertension. Laboratory tests confirmed endogenous hypercortisolism. The MRI revealed a right-sided pituitary microadenoma. The petrosal sinus sampling confirmed the location of the adenoma, and a diagnosis of CD was established. The patient underwent transcranial surgery (mini-pterional craniotomy) with total exeresis of the pituitary gland. The child did not present recurrence.
CASE 4
A 12-year-old female adolescent presented to the outpatient consultation with an illness history of 2.5 years. The father stated that he noticed a short stature for her age, increased body and facial hair, and a round face. He also reported her child was forgetful and tired. On physical examination, the child had cushingoid facies, short stature, hirsutism, and central obesity. Laboratory tests confirmed endogenous hypercortisolism. The MRI showed a microadenoma on the right side and midline, which was later confirmed by petrosal sinus sampling test. Initially, a selective adenomectomy through a transcranial approach was performed. However, due to persistent hypercortisolism, a hypophysectomy through a microscopic transsphenoidal approach was then intended after 6 months. Unfortunately, a neoformation of the intercavernous sinus in the sellar diaphragm provoked abundant bleeding when opening it, and the operation was aborted. A third and definitive procedure was performed through an endoscopic transsphenoidal approach. The patient continues her control queries with the pediatric endocrinologist for postsurgical hypopituitarism.
CASE 5
A 12-year-old male adolescent presented to the outpatient consultation with an illness history of 4 years. The mother stated that she noticed weight gain and short stature. She also reported hyperpigmentation of the skin and acne. Two years earlier, hair began to appear on her face and body. An MRI showed an intrasellar pituitary expansion process, located inferomedially in the adenohypophysis. The patient denied impaired vision and nausea but reported joint pain in the knees, and lumbar region. Laboratory tests revealed endogenous hypercortisolism. Physical examination, confirmed the presence of facial acne, acanthosis nigricans, dry skin, hypertrichosis, and cushingoid facies. At other consultations, the patient reported occasional headache and mild fatigue. He was referred to the Department of Pediatric Neurosurgery for microscopic transsphenoidal surgery. The patient has been following her control queries with the pediatric endocrinologist for 2 years.
DISCUSSION
Clinical presentation
CD has an insidious onset in most children and is usually manifested by gain, lack of height gain, and changes in facial appearance.[
It is important to add that subtle or subclinical presentation is uncommon. Nevertheless, parents and general practitioners fail to identify the pathological changes in the child’s appearance. For this reason, the duration of symptoms until a diagnosis ranges from 0.5 to 3.5 years.[
Regarding a gender-dependent analysis in the context of pediatric CD, contrary to the situation in adults, the distribution between males and females is equal, but male patients have higher body mass index, shorter stature, and higher elevated plasma ACTH levels.[
Diagnosis
The diagnosis of CD is based on clinical suspicion, laboratory tests, and medical imaging. After excluding prolonged exogenous glucocorticoid intake, the first step is to measure cortisol levels by the following tests: 24-hour urinary free cortisol (UFC), dexamethasone suppression test (DST), or late-night salivary/serum cortisol. Two abnormal results are required to confirm hypercortisolism.[
A 24 hour-UFC with a cut-off value of >193 nmoL/day has a sensitivity of 88% in the pediatric population and serial measurements are recommended to increase its accuracy.[
Once the diagnosis of hypercortisolism is established, morning plasma ACTH levels are assessed to determine the etiology. Levels higher than 29 pg/mL suggest ACTH-dependent CS with a sensitivity of 70% and a specificity of 100%, while values <5 pg/mL indicate ACTH-independent CS.[
To localize the pituitary tumor, the standard medical imaging technique is MRI with contrast.[
BPSS is a confirmatory procedure used when MRI does not show a pituitary adenoma, its size is <6 mm or tests are inconclusive.[
Management
CD is a heterogeneous disorder that requires multidisciplinary management by pediatric neurosurgeons, pediatric endocrinologists, and radiologists. Therapeutic options include medical treatment, pituitary surgery, and radiotherapy. As it is a localized tumor, the ideal procedure is surgical resection by selective adenomectomy. Surgical approaches include transcranial surgery, and endoscopic or microscopic transsphenoidal surgery.[
Regarding transsphenoidal surgery, some technical and anatomical aspects, such as pneumatization of the sphenoid sinus and intercarotid distance, should be considered in pediatric patients. First, the surgical corridor is smaller in children due to their smaller nostrils, which makes maneuvering in the posterior fossa more difficult.[
Pituitary radiotherapy is a second-line treatment option following an unsuccessful surgery for pediatric CD and requires combination with pharmacological agents such as ketoconazole.[
Postoperative complications
Between transsphenoidal and transcranial approaches, the latter is associated with more postoperative complications.[
Remission and recurrence
The goal of adenomectomy is remission or cure, although the term “remission” is more appropriate due to the high recurrence rate of CD. At present, there is no consensus on the definition of remission, but the main objectives are reversal of clinical features, normalization of biochemical changes with minimal morbidity, and long-term control without recurrence.[
Post-transsphenoidal surgery remission rate ranges from 70% to 100% over a median time of 7.2 years.[
Long-term outcomes
Each patient’s catch-up growth depends on early identification of growth hormone deficiency and subsequent treatment after surgery or radiotherapy completion. Overall, patients treated with growth hormone therapy and those without the deficiency complication show improvement in standard deviation of height and attainment of target height in adulthood.[
CONCLUSION
CD in children is a heterogeneous disorder that requires multidisciplinary diagnosis and management by pediatric neurosurgeons, pediatric endocrinologists, and radiologists. It should be considered in the differential diagnosis of a child or adolescent who presents mainly with growth failure and obesity. Serial laboratory tests are crucial for diagnosis. Although the preferred imaging to localize the tumor is MRI with contrast, in some cases, no pituitary adenoma could be detected. Then, BPSS gives an accurate localization and lateralization of the adenoma. Regarding treatment, transsphenoidal selective adenomectomy is the optimal treatment due to its higher remission rates. However, technical and anatomical aspects, such as smaller operative corridors, little pneumatization of the sphenoid sinus, and smaller intercarotid distance and piriform distance, should be considered in pediatric patients. A close follow-up of growth, weight, sexual development, and pituitary function parameters is strictly necessary after surgery.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
1. Acharya SV, Gopal RA, Goerge J, Menon PS, Bandgar TR, Shah NS. Radiotherapy in paediatric Cushing’s disease: Efficacy and long term follow up of pituitary function. Pituitary. 2010. 13: 293-7
2. Batista DL, Oldfield EH, Keil MF, Stratakis CA. Postoperative testing to predict recurrent Cushing disease in children. J Clin Endocrinol Metab. 2009. 94: 2757-65
3. Batista DL, Riar J, Keil M, Stratakis CA. Diagnostic tests for children who are referred for the investigation of Cushing syndrome. Pediatrics. 2007. 120: e575-86
4. Biller BM, Grossman AB, Stewart PM, Melmed S, Bertagna X, Bertherat J. Treatment of adrenocorticotropin-dependent Cushing’s syndrome: A consensus statement. J Clin Endocrinol Metab. 2008. 93: 2454-62
5. Bora SK, Suri A, Khadgawat R, Tandon N, Suri V, Chand Sharma M. Management of Cushing’s disease: Changing trend from microscopic to endoscopic surgery. World Neurosurg. 2020. 134: e46-54
6. Castinetti F, Morange I, Conte-Devolx B, Brue T. Cushing’s disease. Orphanet J Rare Dis. 2012. 7: 41
7. Chen S, Chen K, Lu L, Zhang X, Tong A, Pan H. The effects of sampling lateralization on bilateral inferior petrosal sinus sampling and desmopressin stimulation test for pediatric Cushing’s disease. Endocrine. 2019. 63: 582-91
8. Davies JH, Storr HL, Davies K, Monson JP, Besser GM, Afshar F. Final adult height and body mass index after cure of paediatric Cushing’s disease. Clin Endocrinol (Oxf). 2005. 62: 466-72
9. Devoe DJ, Miller WL, Conte FA, Kaplan SL, Grumbach MM, Rosenthal SM. Long-term outcome in children and adolescents after transsphenoidal surgery for Cushing’s disease. J Clin Endocrinol Metab. 1997. 82: 3196-202
10. Ferrigno R, Hasenmajer V, Caiulo S, Minnetti M, Mazzotta P, Storr HL. Paediatric Cushing’s disease: Epidemiology, pathogenesis, clinical management and outcome. Rev Endocr Metab Disord. 2021. 22: 817-35
11. Gazioglu N, Canaz H, Camlar M, Tanrıöver N, Kocer N, Islak C. Neurosurgical treatment of Cushing disease in pediatric patients: Case series and review of literature. Childs Nerv Syst. 2019. 35: 2127-32
12. Gkourogianni A, Sinaii N, Jackson SH, Karageorgiadis AS, Lyssikatos C, Belyavskaya E. Pediatric Cushing disease: Disparities in disease severity and outcomes in the Hispanic and African-American populations. Pediatr Res. 2017. 82: 272-7
13. Grober Y, Grober H, Wintermark M, Jane JA, Oldfield EH. Comparison of MRI techniques for detecting microadenomas in Cushing’s disease. J Neurosurg. 2018. 128: 1051-7
14. Hanba C, Svider PF, Shkoukani MA, Sheyn A, Jacob JT, Eloy JA. Pediatric pituitary resection: Characterizing surgical approaches and complications. Int Forum Allergy Rhinol. 2017. 7: 72-9
15. Ioachimescu AG. Prognostic factors of long-term remission after surgical treatment of Cushing’s disease. Endocrinol Metab Clin North Am. 2018. 47: 335-47
16. Joshi SM, Hewitt RJ, Storr HL, Rezajooi K, Ellamushi H, Grossman AB. Cushing’s disease in children and adolescents: 20 years of experience in a single neurosurgical center. Neurosurgery. 2005. 57: 281-5 discussion 281-5
17. Juszczak A, Ertorer ME, Grossman A. The therapy of Cushing’s disease in adults and children: An update. Horm Metab Res. 2013. 45: 109-17
18. Lebrethon MC, Grossman AB, Afshar F, Plowman PN, Besser GM, Savage MO. Linear growth and final height after treatment for Cushing’s disease in childhood. J Clin Endocrinol Metab. 2000. 85: 3262-5
19. Leinung MC, Kane LA, Scheithauer BW, Carpenter PC, Laws ER, Zimmerman D. Long term follow-up of transsphenoidal surgery for the treatment of Cushing’s disease in childhood. J Clin Endocrinol Metab. 1995. 80: 2475-9
20. Libuit LG, Karageorgiadis AS, Sinaii N, Nguyen May NM, Keil MF, Lodish MB. A gender-dependent analysis of Cushing’s disease in childhood: Pre-and postoperative follow-up. Clin Endocrinol (Oxf). 2015. 83: 72-7
21. Marino AC, Taylor DG, Desai B, Jane JA. Surgery for pediatric pituitary adenomas. Neurosurg Clin N Am. 2019. 30: 465-71
22. Nayak S, Dabadghao P, Dixit P, Dwivedi V, Srivastava AK, Behari S. Cushing’s disease in children: A review. Neurology India. 2020. 68: S52-65
23. Nieman LK, Biller BM, Findling JW, Murad MH, Newell-Price J, Savage MO. Treatment of Cushing’s syndrome: An endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2015. 100: 2807-31
24. Nieman LK, Oldfield EH, Wesley R, Chrousos GP, Loriaux DL, Cutler GB. A simplified morning ovine corticotropin-releasing hormone stimulation test for the differential diagnosis of adrenocorticotropin-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 1993. 77: 1308-12
25. Pandey P, Ojha BK, Mahapatra AK. Pediatric pituitary adenoma: A series of 42 patients. J Clin Neurosci. 2005. 12: 124-7
26. Pasternak-Pietrzak K, Moszczyńska E, Roszkowski M, Szalecki M. Predictive factors for the recurrence of Cushing’s disease after surgical treatment in childhood. Endokrynol Pol. 2020. 71: 313-8
27. Patronas N, Bulakbasi N, Stratakis CA, Lafferty A, Oldfield EH, Doppman J. Spoiled gradient recalled acquisition in the steady state technique is superior to conventional postcontrast spin echo technique for magnetic resonance imaging detection of adrenocorticotropin-secreting pituitary tumors. J Clin Endocrinol Metab. 2003. 88: 1565-9
28. Savage MO, Chan LF, Grossman AB, Storr HL. Work-up and management of paediatric Cushing’s syndrome. Curr Opin Endocrinol Diabetes Obes. 2008. 15: 346-51
29. Shah NS, George J, Acharya SV, Lila AR, Sarathi V, Bandgar TR. Cushing disease in children and adolescents: Twenty years’ experience in a tertiary care center in India. Endocr Pract. 2011. 17: 369-76
30. Storr HL, Chan LF, Grossman AB, Savage MO. Paediatric Cushing’s syndrome: Epidemiology, investigation and therapeutic advances. Trends Endocrinol Metab. 2007. 18: 167-74
31. Storr HL, Plowman PN, Carroll PV, François I, Krassas GE, Afshar F. Clinical and endocrine responses to pituitary radiotherapy in pediatric Cushing’s disease: An effective second-line treatment. J Clin Endocrinol Metab. 2003. 88: 34-7
32. Storr HL, Savage MO. Management of endocrine disease: Paediatric Cushing’s disease. Eur J Endocrinol. 2015. 173: R35-45
33. Stratakis CA. An update on Cushing syndrome in pediatrics. Ann Endocrinol (Paris). 2018. 79: 125-31
34. Tatreau JR, Patel MR, Shah RN, McKinney KA, Zanation AM. Anatomical limitations for endoscopic endonasal skull base surgery in pediatric patients. Laryngoscope. 2010. 120: S229
35. Tatsi C, Bompou ME, Flippo C, Keil M, Chittiboina P, Stratakis CA. Paediatric patients with Cushing disease and negative pituitary MRI have a higher risk of nonremission after transsphenoidal surgery. Clin Endocrinol (Oxf). 2021. 95: 856-62
36. Tatsi C, Neely M, Flippo C, Bompou ME, Keil M, Stratakis CA. Recovery of hypothalamic-pituitary-adrenal axis in paediatric Cushing disease. Clin Endocrinol (Oxf). 2021. 94: 40-7
37. Turan H, Çatlı G, Kardelen AD, Böber E, Akıncı A, Çetinkaya S. Diagnostic value of bilateral petrosal sinus sampling in children with Cushing disease: A multi-center study. J Clin Res Pediatr Endocrinol. 2022. 14: 29-36
38. Yordanova G, Martin L, Afshar F, Sabin I, Alusi G, Plowman NP. Long-term outcomes of children treated for Cushing’s disease: A single center experience. Pituitary. 2016. 19: 612-24

