

- Department of Neurosurgery, Beneficência Portuguesa Ribeirão Preto, Ribeirão Preto, Brazil.
Correspondence Address:
Joaquim Fechine de Alencar Neto, Department of Neurosurgery, Beneficência Portuguesa Ribeirão Preto, Ribeirão Preto, Brazil.
DOI:10.25259/SNI_338_2023
Copyright: © 2023 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Breno Nery, Joaquim Fechine de Alencar Neto, Layssa Rhossana dos Santos Melo, Rodrigo Antônio Fernandes Costa, Eduardo Quaggio, Luísa Segato de Medeiros, José Alencar de Sousa Segundo, Nicolle Fortuny de Lima, Renan Lopez Rivero. Olfactory groove monophasic sinovial sarcoma and von Recklinghausen’s disease: A case report and literature review. 07-Jul-2023;14:231
How to cite this URL: Breno Nery, Joaquim Fechine de Alencar Neto, Layssa Rhossana dos Santos Melo, Rodrigo Antônio Fernandes Costa, Eduardo Quaggio, Luísa Segato de Medeiros, José Alencar de Sousa Segundo, Nicolle Fortuny de Lima, Renan Lopez Rivero. Olfactory groove monophasic sinovial sarcoma and von Recklinghausen’s disease: A case report and literature review. 07-Jul-2023;14:231. Available from: https://surgicalneurologyint.com/surgicalint-articles/12400/
Date of Submission
18-Apr-2023
Date of Acceptance
13-Jun-2023
Date of Web Publication
07-Jul-2023
Abstract
Background: Soft-tissue sarcomas are a rare and diverse group of neoplastic lesions. They represent only 1% of malignant tumors in adults and 15% in children. Synovial sarcoma (SS) is a type of soft-tissue sarcoma, accounting for 5–10% of cases, and commonly affecting extremities. Diagnosis, treatment, and prognosis remain challenging especially when localized in uncommon areas, such as intracranial lesions.
Case Description: A 13-year-old male patient with a clinical history of neurofibromatosis Type I (NF1) presenting holocranial headache with jet vomiting and apathy 2 days before admission, without neurological deficits and/or focal findings. On magnetic resonance imaging: an extra-axial infiltrative lesion with contrast uptake at the base of the skull in the olfactory groove topography. After total tumor resection, the anatomopathological examination showed monophasic SS. The patient returned after 6 months with similar symptoms, and the lesion recurred and was reoperated. Unfortunately, 7 months after the second surgery, the patient died.
Conclusion: SS can occur extraarticulously and with a variable clinical presentation and poor prognosis despite adjuvant therapies with radiotherapy and chemotherapy. In individuals with clinical history of NF1, there is still no direct correlation between the two manifestations, although current descriptions are suggestive of a possible interaction.
Keywords: Case report, Intracranial, Monophasic synovial sarcoma, Neurofibromatosis Type 1
INTRODUCTION
Soft-tissue sarcoma comprises a group of rare neoplastic lesions with mesenchymal origin, with about 60 distinct possible diagnoses within this group of tumors. In addition to the complexity and variety of lesions described, this set represents only 1% of malignant tumors in adults and about 15% in children.[
Cases of metastatic intracranial SS are well described in the literature, and one of the possible sites of neoplastic dissemination is the central nervous system.[
MATERIALS AND METHODS
The article presents a case report with a review of the literature. The literature review focuses on case reports of patients with primary intracranial SS. The selection of articles was based on an analysis of the PubMed database using the following keywords: “synovial sarcoma,” “soft-tissue sarcoma,” and “intracranial synovial sarcoma.” To extend the search, the Boolean operators “AND” and “OR” were used to filter articles that were relevant to the study. No filter was used during the search or year limitations were imposed. The search was conducted by one of the researchers using advanced search resources provided by PubMed. To increase the number of publications, the search terms were crossed. The inclusion criteria were limited to case report articles about primary intracranial SS, articles of intracranial metastasis and other types of soft-tissue sarcoma were excluded from the study. Fifteen articles met the inclusion criteria and were used to compose the
CASE REPORT
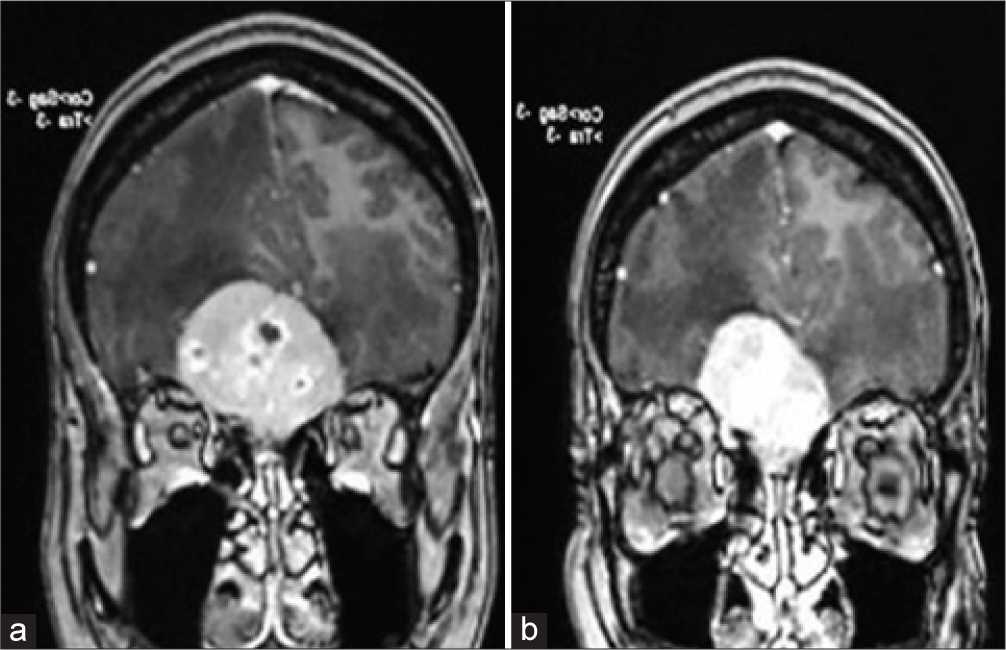
A 13-year-old male patient with a clinical history of NF1 was admitted from work presenting with a holocranial headache associated with jet vomiting and significant apathy for 2 days before admission. On physical examination, there was no neurological deficits or focal findings. On inspection, the presence of multiple café-au-lait spots, at least 6, and several dermal neurofibromas were identified, corroborating the previous diagnosis of NF1. On complementary magnetic resonance imaging investigation, a 4.0 × 3.8 cm infiltrative extra-axial lesion was observed, with contrast uptake, located at the base of the skull in the olfactory groove topography associated with intense peritumoral edema and absence of other lesions [
Through clinical findings, the surgical approach was adopted through bifrontal craniotomy and interhemispheric subfrontal approach for tumor resection. A spongy, extradural infiltrative lesion in the brain parenchyma and skull base was visualized. After gross total resection, the pathological examination showed MSS in the olfactory groove [
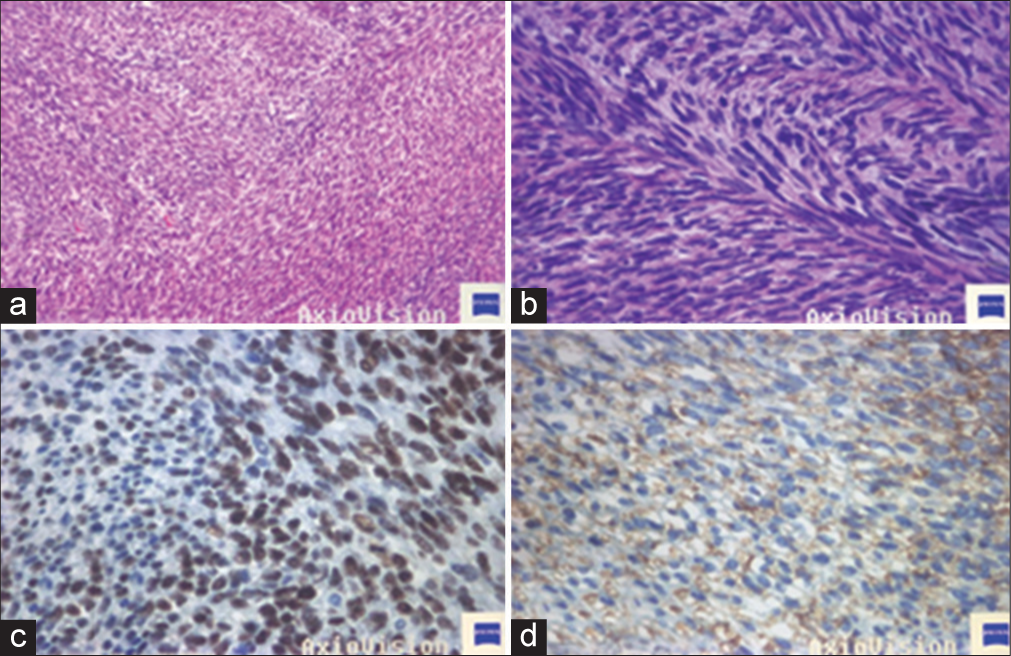
Figure 3:
(a) 100XX - Monophasic synovial sarcoma with fibrosarcoma-like areas, spindle-shaped cells with moderate nuclear pleomorphism arranged in long and intercalated fascicles. (b) 400 XX – Hyperchromatic, fusiform nuclei cells arranged in long bundles. (c) TLE-1 400XX – Strong and diffuse nuclear expression antibody in neoplasia. (d) CD99 400XX – Cytoplasmatic and diffuse membrane expression antibody.
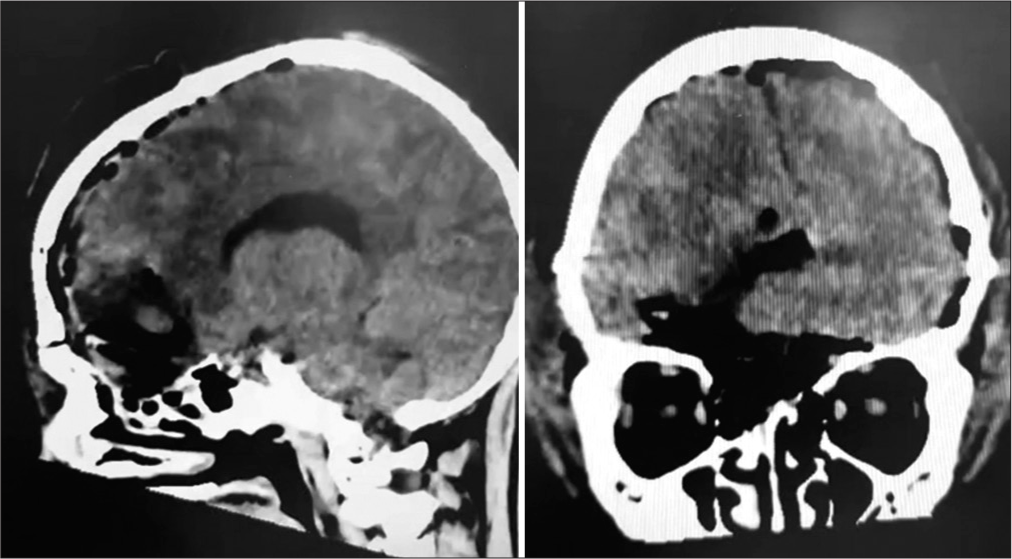

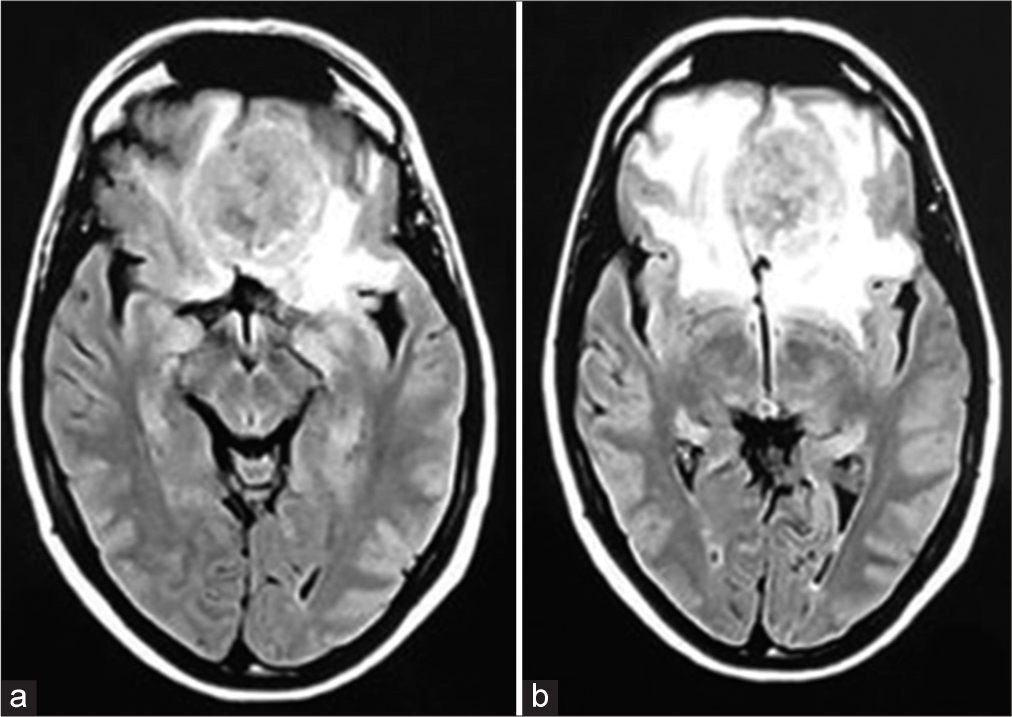
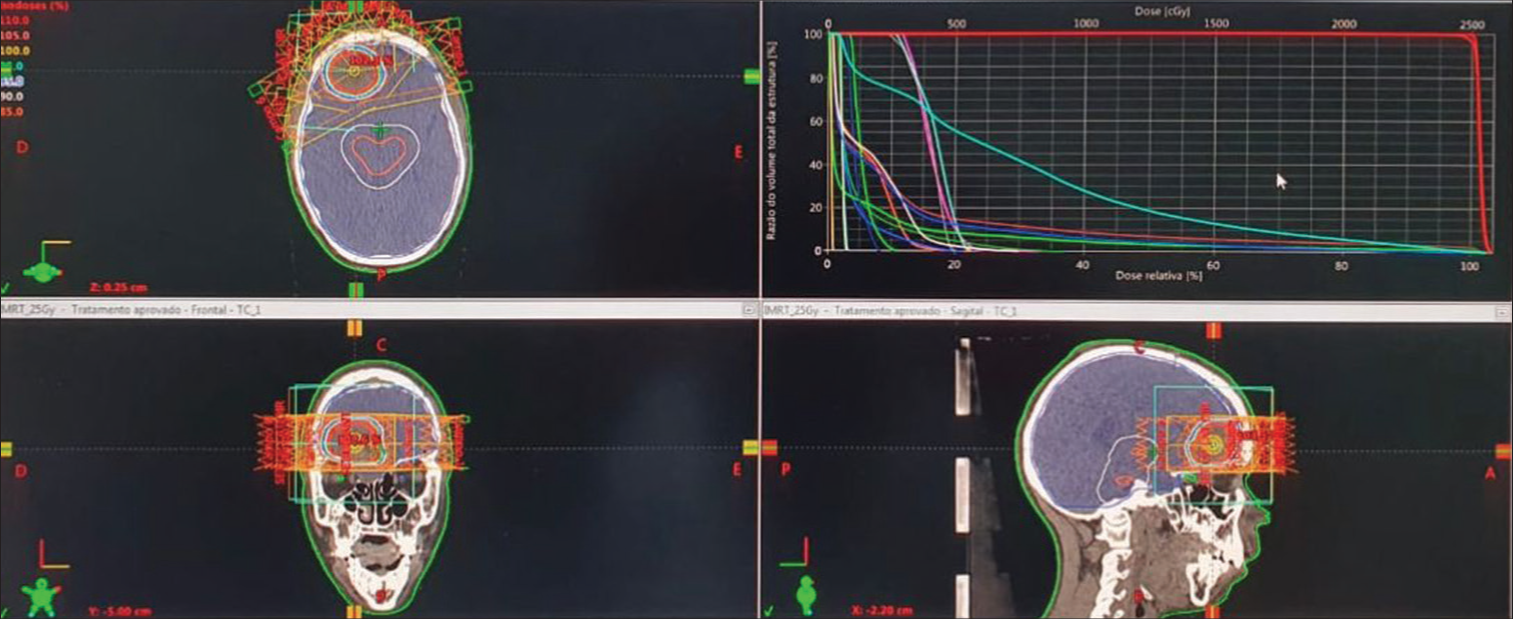
The patient returned to the hospital with similar symptoms to the first diagnosis (5 months after the procedure), evaluations showed that the lesion had relapsed and hence was reoperated on, in which the MSS was again gross totally resected, and the patient was further referred for oncologic adjuvant treatment with fractionated stereotactic radiotherapy [
DISCUSSION
Soft-tissue tumors comprise a broad and diverse group of lesions with distinct clinical characteristics and presentations that differ from mesenchymal tissue, being neoplasms that may present in different age groups, as well as variable topography, prognosis, treatments, and histopathological analysis.[
The clinical presentation of this condition varies according to topography, and the extremities, joints, or bursa are the most frequent sites of involvement, although the temporomandibular joint and knee are the most injured.[
Clinical and surgical management
Among all 30 cases collected in this article, of which 14 are described in
Laboratory diagnosis and analysis
In general, the macroscopic analysis of the lesion is evident that it is a heterogeneous, multinodular mass, well adhered to adjacent planes, infiltrative, and irregular. In addition, intracranial lesions present with a high aggressiveness and a rapid decline in neurological function, and dural adherence is described in most cases reported in the literature.[
Some markers are essential for identification and differentiation of lesion types. Cytokeratin CK19 And CK7, EMA, BCL-2, CD34, CD99, S-100, and transducer-like enhancer of split 1 are some useful examples for the identification and differentiation of SS from other lesions.[
Correlation with NF1
NF1 is a disease of autosomal dominant genetic origin characterized by the mutation of the NF1 gene on chromosome 17q11.2. It was first described by Frederick von Recklinghausen and whose responsible gene was identified in 1990.[
Although the typical description of the NF1 gene mutation is linked to changes in the peripheral nervous system, there are descriptions of NF1 alterations related to the generation of lesions of the soft-tissue tumor group, such as pleomorphic liposarcomas myxofibrosarcomas, lesions of sarcomas subtypes.[
CONCLUSION
The present case report is an unusual intracranial presentation of primary MSS, associated with a patient with NF1 disease. It is a rare condition described in the literature, with complex diagnosis and essentially surgical treatment, with adjuvant radiotherapy alone or associated chemotherapy. The prognosis of these patients is poor, with reduced survival and several reports of recurrence. Despite the description of other cases that correlate the NF1 gene mutation and the presence of soft-tissue sarcomas, the description of cases correlating SSs and von Recklinghausen’s disease is still incipient and further investigation should be performed to properly address its relationship.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
1. Aggad M, Gkasdaris G, Rousselot C, Destrieux C, François P, Velut S. Intracranial primary synovial sarcoma mimicking a spontaneous cerebral hematoma-a case report and review of the literature. Neurochirurgie. 2022. 68: 443-6
2. Akdeniz N. Rare occurrence of synovial sarcoma originating from dura mater. Turk J Oncol. 2019. 34: 52-5
3. Ayodele O, Razak AR. Immunotherapy in soft-tissue sarcoma. Curr Oncol. 2020. 27: 17-23
4. Barretina J, Taylor BS, Banerji S, Ramos AH, Lagos-Quintana M, Decarolis PL. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010. 42: 715-21
5. Chan JA, McMenamin ME, Fletcher CD. Synovial sarcoma in older patients: Clinicopathological analysis of 32 cases with emphasis on unusual histological features. Histopathology. 2003. 43: 72-83
6. Crew AJ, Clark J, Fisher C, Gill S, Grimer R, Chand A. Fusion of SYT to two genes, SSX1 and SSX2, encoding proteins with homology to the Kruppel-associated box in human synovial sarcoma. EMBO J. 1995. 14: 2333-40
7. Dodd RD, Mito JK, Eward WC, Chitalia R, Sachdeva M, Ma Y. NF1 deletion generates multiple subtypes of soft-tissue sarcoma that respond to MEK inhibition. Mol Cancer Ther. 2013. 12: 1906-17
8. Ewing CA, Zakowski MF, Lin O. Monophasic synovial sarcoma: A cytologic spectrum. Cytopathol Diagn. 2004. 30: 19-23
9. Fisher C. Synovial sarcoma. Ann Diagn Pathol. 1998. 2: 401-21
10. Friedman JM. Epidemiology of neurofibromatosis Type 1. Am J Med Genet. 1999. 89: 1-6
11. Friedman JM. Neurofibromatosis 1: Clinical manifestations and diagnostic criteria. J Child Neurol. 2002. 17: 548-54 discussion 571-2, 646-51
12. Horbinski C, Cieply K, Bejjani GK, McFadden K. Primary intracranial dural-based synovial sarcoma with an unusual SYT fluorescence in situ hybridization pattern. J Neurosurg. 2008. 109: 897-903
13. Jagdis A, Rubin BP, Tubbs RR, Pacheco M, Nielsen TO. Prospective evaluation of TLE1 as a diagnostic immunohistochemical marker in synovial sarcoma. Am J Surg Pathol. 2009. 33: 1743-51
14. Katsaros VK, Katsarou AA, Papadopoulou A, Floros D, Marangos N. Intracranial primary synovial sarcoma: Radiologicpathologic correlation. Neuroradiol J. 2008. 21: 362-7
15. Kleinschmidt-DeMasters BK, Mierau GW, Sze CI, Breeze RE, Greffe B, Lillehei KO. Unusual dural and skull-based mesenchymal neoplasms: A report of four cases. Hum Pathol. 1998. 29: 240-5
16. Lin YJ, Yang QX, Tian XY, Li B, Li Z. Unusual primary intracranial dural-based poorly differentiated synovial sarcoma with t(X; 18)(p11; q11). Neuropathology. 2013. 33: 75-82
17. Manizhe AK, Mohseni I, Sahranavard A, Tabrizi Z. Recurrent primary intracranial synovial sarcoma, a case report and review of the literature. Clin Case Rep. 2022. 10: e6273
18. McCool A, Turner C, Turner S, Heppner P, Saran F. Primary intraventricular synovial sarcoma of the brain with recurrence-case presentation. BMC Neurol. 2022. 22: 447
19. Olsen SH, Thomas DG, Lucas DR. Cluster analysis of immunohistochemical profiles in synovial sarcoma, malignant peripheral nerve sheath tumor, and Ewing sarcoma. Pathol Mod. 2006. 19: 659-68
20. Otani Y, Ichikawa T, Kurozumi K, Yanai H, Kunisada T, Ozaki T. A case of synovial sarcoma with brain metastasis treated with surgical resection and stereotactic radiosurgery. No Shinkei Geka. 2013. 41: 255-62
21. Patel M, Li L, Nguyen HS, Doan N, Sinson G, Mueller W. Primary intracranial synovial sarcoma. Case Rep Neurol Med. 2016. 2016: 5608315
22. Scheithauer BW, Silva AI, Kattner K, Seibly J, Oliveira AM, Kovacs K. Synovial sarcoma of the sellar region. Neuro Oncol. 2007. 9: 454-9
23. Sharma S, Sharma A, Lobo G, Nayak M, Pradhan D, Samriti . Primary dura-based synovial sarcoma of the parafalcine region of brain. Pathol Res Pract. 2017. 213: 868-71
24. Shi W, Indelicato DJ, Morris CG, Scarborough MT, Gibbs CP, Zlotecki RA. Long-term treatment outcomes for patients with synovial sarcoma: A 40-year experience at the University of Florida. Am J Clin Oncol. 2013. 36: 83-8
25. Singh HP, Grover S, Garg B, Sood N. Histopathological spectrum of soft-tissue tumors with immunohistochemistry correlation and FNCLCC grading: A North Indian experience. Niger Med J. 2017. 58: 149-55
26. Skytting B, Nilsson G, Brodin B, Xie Y, Lundeberg J, Uhlén M. A novel fusion gene, SYT-SSX4, in synovial sarcoma. J Natl Cancer Inst. 1999. 91: 974-5
27. Stacchiotti S, Van Tine BA. Synovial sarcoma: Current concepts and future perspectives. J Clin Oncol. 2018. 36: 180-7
28. Thway K, Fisher C. Synovial sarcoma: Defining features and diagnostic evolution. Ann Diagn Pathol. 2014. 18: 369-80
29. Vergara-Lluri ME, Stohr BA, Puligandla B, Brenholz P, Horvai AE. A novel sarcoma with dual differentiation: Clinicopathologic and molecular characterization of a combined synovial sarcoma and extraskeletal myxoid chondrosarcoma. Am J Surg Pathol. 2012. 36: 1093-8
30. Von Mehren M, Kane JM, Agulnik M, Bui MM, Carr-Ascher J, Choy E. Soft tissue sarcoma, version 2.2022, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2022. 20: 815-33
31. Vora TK, Lath R, Swain M, Ray A. Primary intracranial synovial sarcoma: A case report and review of literature. Surg Neurol Int. 2022. 13: 447
32. Wang YY, Li ML, Zhang ZY, Ding JW, Xiao LF, Li WC. Primary intracranial synovial sarcoma with hemorrhage: A case report. World J Clin Cases. 2021. 9: 8871-8
33. Ward BA, Gutmann DH. Neurofibromatosis 1: From lab bench to clinic. Pediatr Neurol. 2005. 32: 221-8
34. Wrba F, Fertl H, Amann G, Tell E, Krepler R. Epithelial markers in synovial sarcoma. An immunohistochemical study on paraffin embedded tissues. Virchows Arch A Pathol Anat Histopathol. 1989. 415: 253-8
35. Xiao GY, Pan BC, Tian XY, Li Y, Li B, Li Z. Synovial sarcoma in cerebellum: A case report and literature review. Brain Tumor Pathol. 2014. 31: 68-75
36. Zhang G, Xiao B, Huang H, Zhang Y, Zhang X, Zhang J. Intracranial synovial sarcoma: A clinical, radiological and pathological study of 16 cases. Eur J Surg Oncol. 2019. 45: 2379-85

