

- Department of Pathology and Cell Biology, Columbia University Irving Medical Center, New York, United States.
- Department of Neurosurgery, Columbia University Irving Medical Center, New York, United States.
- Department of Pathology and Laboratory Medicine, Dartmouth-Hitchcock Medical Center, Lebanon, New Hampshire, United States.
- Norris Cotton Cancer Center, Dartmouth-Hitchcock Medical Center, Lebanon, New Hampshire, United States.
Correspondence Address:
George Zanazzi, Department of Pathology and Laboratory Medicine, Dartmouth-Hitchcock Medical Center, New Hampshire, United States.
DOI:10.25259/SNI_443_2022
Copyright: © 2022 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Osama A. Al-Dalahmah1, Linda Wang2, Susan J. Hsiao1, Chun-Chieh Lin3, Mahesh M. Mansukhani1, Peter Canoll1, Jeffrey N. Bruce2, George Zanazzi3,4. Pineal region ganglioglioma: A neoplasm with a bimodal age distribution. 10-Jun-2022;13:245
How to cite this URL: Osama A. Al-Dalahmah1, Linda Wang2, Susan J. Hsiao1, Chun-Chieh Lin3, Mahesh M. Mansukhani1, Peter Canoll1, Jeffrey N. Bruce2, George Zanazzi3,4. Pineal region ganglioglioma: A neoplasm with a bimodal age distribution. 10-Jun-2022;13:245. Available from: https://surgicalneurologyint.com/surgicalint-articles/11646/
Date of Submission
10-May-2022
Date of Acceptance
19-May-2022
Date of Web Publication
10-Jun-2022
Abstract
Background: Gangliogliomas arise very rarely in the pineal region, where their natural histories and pathologic features are poorly understood.
Case Description: In this report, we describe a 36-year-old woman who presented with a seizure followed by worsening headache, dizziness, confusion, and intermittent left facial numbness over the next few weeks. A head CT scan showed a partially calcified pineal region mass with hydrocephalus. After an endoscopic third ventriculostomy, the patient underwent a resection of the tumor that contained dysplastic ganglion cells and piloid glial cells. Molecular profiling of this CNS WHO Grade 1 ganglioglioma revealed polysomies of chromosomes 7 and 9, and a BUB1 variant of uncertain significance, without known MAP kinase pathway alterations. From a review of the literature, we found two distinct age distributions for pineal ganglioglioma, with modes at 1 and 36 years of age.
Conclusion: Although very rare, this tumor should be considered in the differential diagnosis of pineal region tumors in children and young adults.
Keywords: Epiphysis, Glioneuronal, Targeted next-generation sequencing, Tumor, Chromosome 7 polysomy
INTRODUCTION
Pineal region tumors constitute 2.8% of intracranial tumors in children and adolescents in the United States, and 0.05% in adults.[
Gangliogliomas are tumors characterized by dysplastic ganglion cells and neoplastic glial cells that most commonly occur in children and young adults. Together with gangliocytomas, they comprise 0.4% of all central nervous system tumors. While gangliogliomas can be found throughout the central nervous system, from the cerebral hemispheres to the spinal cord, most tumors occur in the temporal lobe and present with focal seizures. Complete surgical resection of these well-circumscribed, variably cystic lesions leads to an excellent prognosis for most of these patients, and hence they correspond to CNS WHO Grade 1. A small subset of these tumors show aggressive behavior and anaplastic histologic features, which correspond to CNS WHO grade 3 (reviewed in WHO Classification of Tumours Editorial Board[
Although a ganglioglioma in the pineal region was first described in 1928,[
CASE HISTORY
The patient is a 36-year-old right-handed woman who collapsed and was found unresponsive, stiff, drooling, and tremulous, but with no rhythmic shaking, tongue-biting, or urinary incontinence. She became responsive again within 4–5 min. Head CT and subsequent MRI at an outside hospital revealed a mass in the pineal region. The patient was started on dexamethasone and levetiracetam.
She experienced worsening headache, dizziness, confusion, and intermittent left facial numbness. Head CT revealed a partially calcified mass centered in the pineal region with moderate lateral ventricle and third ventricle hydrocephalus. An endoscopic third ventriculostomy was performed. Headache, left-sided facial numbness, and confusion diminished. Serum AFP and beta-HCG levels were normal. Brain MRI showed a 3.0 × 2.1 cm heterogeneously enhancing mass in the pineal region [
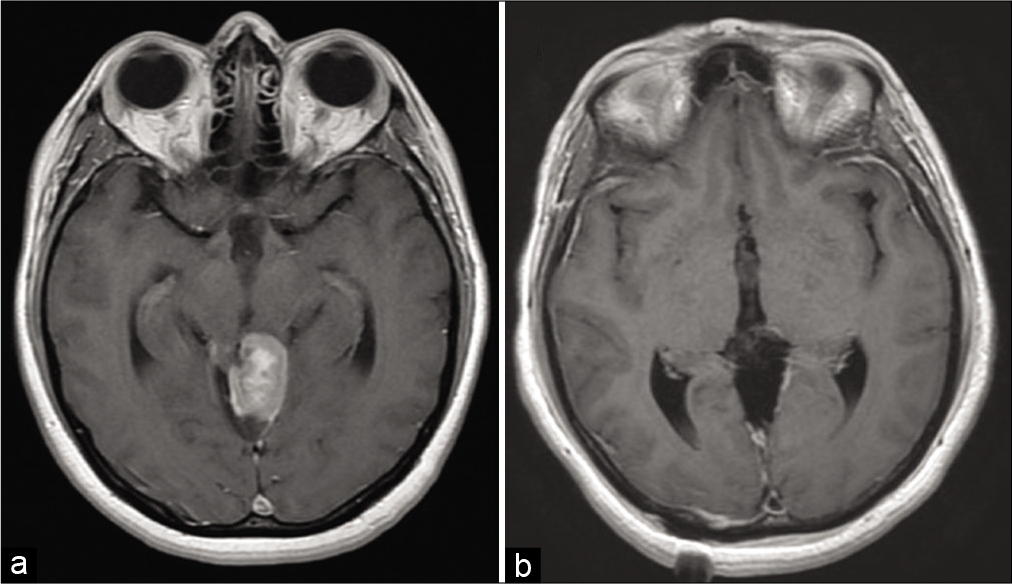
Figure 1:
Preoperative and postoperative imaging of the pineal region neoplasm. (a) Axial T1-weighted MRI shows a mass, measuring 3.0 × 2.1 cm, at the level of the pineal gland with heterogeneous, mostly solid enhancement. (b) Axial T1-weighted MRI after resection reveals minimal linear enhancement along the margin of the surgical cavity.
Two and a half weeks later, the patient underwent a right parietal craniotomy followed by an interhemispheric approach to the tumor. The posterior surface of the tumor was partially attached to the cerebellum, and the right lateral aspect of the tumor seemed to blend into the dorsal midbrain. A gross total resection of the tumor was performed [
Pathology
Multiple fragments of tan-gray and red soft tissue were received. Hematoxylin and eosin-stained sections of the resection specimen showed highly pleomorphic tumor cells, with large ganglioid cells containing vesicular nuclei and distinct nucleoli interspersed between variably sized ovoid cells and spindle-shaped bipolar cells containing fibrillary, eosinophilic cytoplasmic processes [
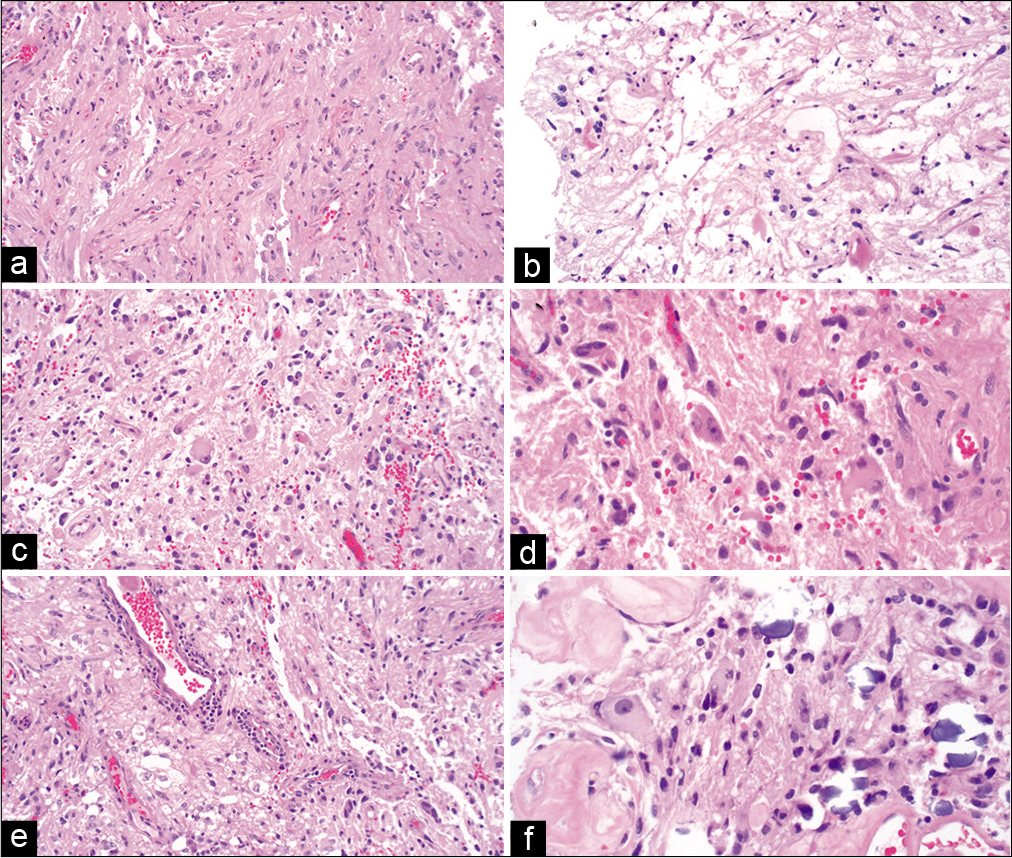
Figure 2:
Histologic features of the pineal region neoplasm. (a) Hematoxylin and eosin-stained sections of the resection specimen show a pleomorphic neoplasm with thick, fibrillary, cytoplasmic processes. Some cells have ovoid nuclei, and some have spindle-shaped nuclei. (b) Eosinophilic granular bodies and Rosenthal fibers are noted on a frozen section. Low magnification (c) and high magnification (d) photomicrographs reveal many ganglioid cells with prominent nucleoli. (e) Collections of mixed chronic inflammatory cells can be seen around some blood vessels. (f) Tumor cells are present adjacent to hyalinized blood vessels and corpora arenacea.
Glial fibrillary acidic protein (GFAP) immunoreactivity was diffusely positive in the glial component [
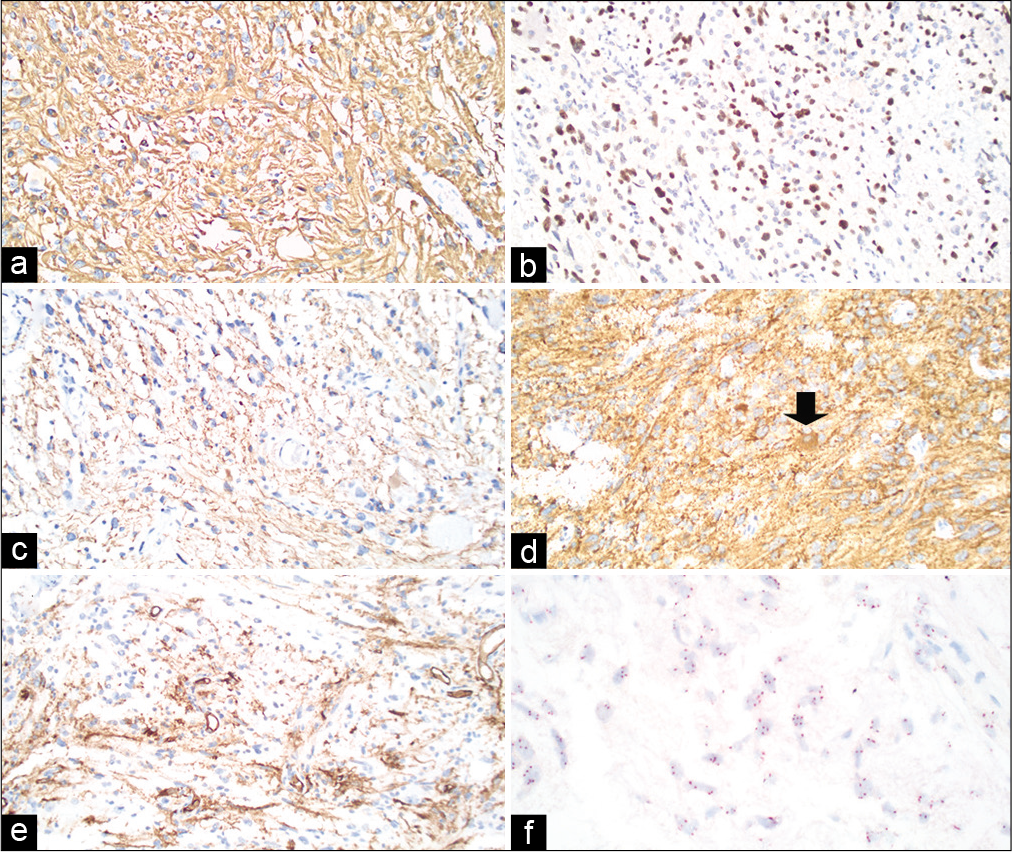
Figure 3:
Immunophenotype and EGFR gene copy number of the pineal region neoplasm. (a) GFAP immunoreactivity is diffusely positive in the glial component. (b) Olig2 is expressed in the majority of cells. (c) A phosphorylated neurofilament immunostain highlights many tumor cell axons. (d) Granular synaptophysin immunoreactivity, including in the soma and axons of a gangliocytic tumor cell (arrow). (e) CD34 is positive in a subset of tumor cells with highly branched morphology and in blood vessels. (f) Evaluation of EGFR (black) and chromosome 7 (CEP7 probe, red) through silver in situ hybridization shows chromosome 7 polysomy without selective EGFR amplification.
Molecular diagnostics
To begin to evaluate the molecular alterations in this tumor, paraffin-embedded sections were incubated with chromosome 7 reference alpha-centromeric (CEP7, red) and EGFR (black) in situ hybridization DNA probes [
Targeted next generation sequencing was performed with the Columbia Combined Cancer Panel, a 467-gene panel.[
DISCUSSION
We present a report of ganglioglioma, atypically located in the pineal region, in a 36-year-old woman who presented with a seizure followed by worsening headache, dizziness, confusion, and intermittent left facial numbness. CT and MRI showed a large, heterogeneously enhancing mass in the pineal region causing obstruction of the aqueduct of Sylvius and hydrocephalus. Following a right parietal craniotomy with interhemispheric approach for tumor resection, examination of the tumor revealed a ganglioglioma, CNS WHO Grade 1, with polysomies of chromosomes 7 and 9 and a BUB1 variant of uncertain significance. Postoperative MRI showed gross total resection. We reviewed our large institutional cohort of 221 patients with pineal region tumors that were resected over a 25 year-period from 1994 to 2019, and this was the only ganglioglioma.[
We reviewed the literature and found 24 additional cases of ganglioglioma in the pineal region [
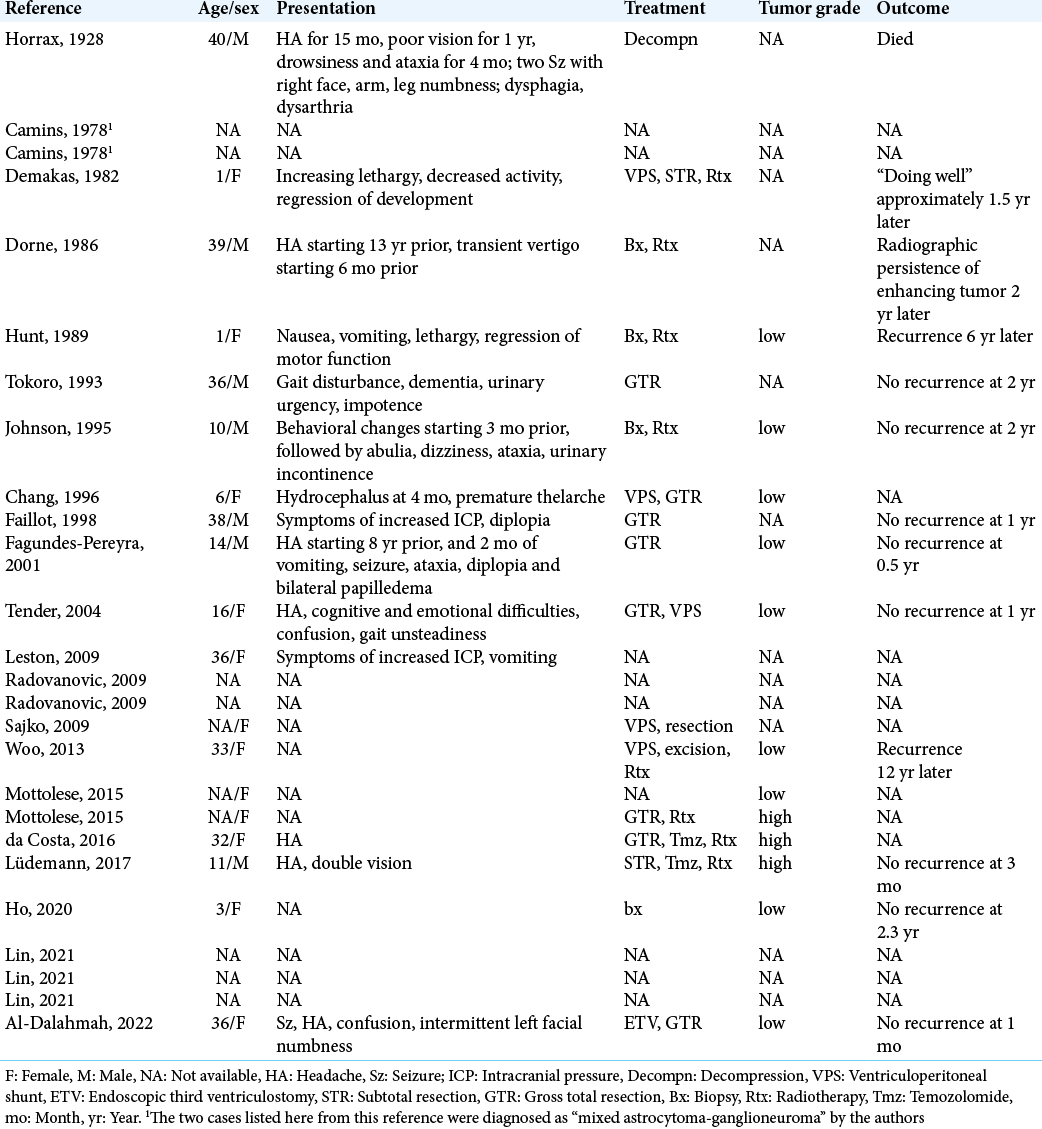
We found that pineal gangliogliomas, like other pineal region lesions, tend to present with signs and symptoms of increased intracranial pressure due to compression of the aqueduct of Sylvius, resulting in headache, nausea, vomiting, and papilledema. Features associated with Parinaud syndrome are also commonly seen, including vertical gaze palsy resulting in diplopia and pupillary light-near dissociation. In the case of our patient, the clinical presentation was also notable for a seizure, a common presentation of gangliogliomas in general due to their typical location in the temporal lobe, but an uncommon presentation for most lesions of the pineal region including gangliogliomas. There may be a higher prevalence of epileptic seizures in some patients with pineal cysts,[
Pineal gangliogliomas, like other gangliogliomas, are characterized by dysplastic neurons and intermixed glia, with immunohistochemical staining positive for markers of both cell lines, such as neuron-specific enolase, neurofilament, synaptophysin, and GFAP. The immunophenotype of the pineal ganglioglioma in our case was overall similar to that of general non-pineal gangliogliomas, with diffusely positive staining for GFAP in the glial component of the tumor [
The genetic features of pineal gangliogliomas are not known, but the most common chromosomal abnormality found in non-pineal gangliogliomas is gain of chromosome 7,[
In general, pineal gangliogliomas are low-grade tumors most often corresponding to CNS WHO Grade 1, similar to gangliogliomas found elsewhere in the brain.[
As with other gangliogliomas throughout the central nervous system, pineal gangliogliomas appear to recur at a low rate. Almost all cases reviewed here were of patients with primary tumors presenting for the 1st time, and surgical resection was curative in most cases. Four patients represented after initial treatment with residual or recurrent disease.[
CONCLUSION
We present a case of a rare pineal region ganglioglioma in an adult woman. Review of our large institutional cohort of 221 pineal region tumors, resected over 25 years, did not reveal any additional gangliogliomas.[
Ethics approval
All procedures performed in this study involving human participants were in accordance with the ethical standards of the research committee of University of Helsinki and with the 1964 Declaration of Helsinki and its amendments or comparable ethical standards.
Declaration of patient consent
Patient’s consent not required as patient’s identity is not disclosed or compromised.
Financial support and sponsorship
Departmental funds.
Conflicts of interest
There are no conflicts of interest.
References
1. Al-Hussaini M, Sultan I, Abuirmileh N, Jaradat I, Qaddoumi I. Pineal gland tumors: Experience from the SEER database. J Neurooncol. 2009. 94: 351-8
2. Burres KP, Hamilton RD. Pineal apoplexy. Neurosurgery. 1979. 4: 264-8
3. Camins MB, Schlesinger EB. Treatment of tumours of the posterior part of the third ventricle and the pineal region: A long term follow-up. Acta Neurochir (Wien). 1978. 40: 131-43
4. Chang YL, Lin SZ, Chiang YH, Liu MY, Lee WH. Pineal ganglioglioma with premature thelarche. Report of a case and review of the literature. Childs Nerv Syst. 1996. 12: 103-6
5. Da Costa MD, Centeno R, Neto M, Dória-Netto H, Filho J, de Araujo Paz D. Anaplastic ganglioglioma of the pineal region-a case report. Arq Bras Neurocir. 2015. 35: 253-6
6. Demakas JJ, Sonntag VK, Kaplan AM, Kelley JJ, Waggener JD. Surgical management of pineal area tumors in early childhood. Surg Neurol. 1982. 17: 435-40
7. Dorne HL, O’Gorman AM, Melanson D. Computed tomography of intracranial gangliogliomas. AJNR Am J Neuroradiol. 1986. 7: 281-5
8. Ebina K, Suzuki S, Takahashi T, Iwabuchi T, Takei Y. Gangliocytoma of the pineal body. A case report and review of the literature. Acta Neurochir (Wien). 1985. 74: 134-40
9. Fagundes-Pereyra WJ, Sousa Ld, Carvalho GT, Sousa AA. Ganglioglioma of the pineal region: Case report. Arq Neuropsiquiatr. 2001. 59: 599-604
10. Faillot T, Sichez JP, Capelle L, Kujas M, Fohanno D. Ganglioglioma of the pineal region: Case report and review of the literature. Surg Neurol. 1998. 49: 104-7
11. Hajnsek S, Paladino J, Gadze ZP, Nanković S, Mrak G, Lupret V. Clinical and neurophysiological changes in patients with pineal region expansions. Coll Antropol. 2013. 37: 35-40
12. Herrick MK, Rubinstein LJ. The cytological differentiating potential of pineal parenchymal neoplasms (true pinealomas). A clinicopathological study of 28 tumours. Brain. 1979. 102: 289-320
13. Ho CY, Bornhorst M, Almira-Suarez MI, Donev K, Grafe M, Gordish-Dressman H. Clinicopathologic features of diencephalic neuronal and glioneuronal tumors. J Neuropathol Exp Neurol. 2020. 79: 67-73
14. Horrax G, Bailey P. Pineal pathology: Further studies. Arch Neurol Psych. 1928. 19: 394-414
15. Hunt SJ, Johnson PC. Melanotic ganglioglioma of the pineal region. Acta Neuropathol. 1989. 79: 222-5
16. Johnson MD, Jennings MT, Lavin P, Creasy JL, Maciunas RJ. Ganglioglioma of the pineal gland: Clinical and radiographic response to stereotactic radiosurgical ablation. J Child Neurol. 1995. 10: 247-9
17. Koelsche C, Wöhrer A, Jeibmann A, Schittenhelm J, Schindler G, Preusser M. Mutant BRAF V600E protein in ganglioglioma is predominantly expressed by neuronal tumor cells. Acta Neuropathol. 2013. 125: 891-900
18. Lang FF, Epstein FJ, Ransohoff J, Allen JC, Wisoff J, Abbott IR. Central nervous system gangliogliomas Part. 2 Clinical outcome. J Neurosurg. 1993. 79: 867-73
19. Leston J, Mottolese C, Champier J, Jouvet A, Brun J, Sindou M. Contribution of the daily melatonin profile to diagnosis of tumors of the pineal region. J Neurooncol. 2009. 93: 387-94
20. Lin CC, Mansukhani MM, Bruce JN, Canoll P, Zanazzi G. Rosette-forming glioneuronal tumor in the pineal region: A series of 6 cases and literature review. J Neuropathol Exp Neurol. 2021. 80: 933-43
21. Lin X, Huang R, Zhang P, Sun J, Dong G, Huang Y. Low-grade gangliogliomas in adults: A population-based study. Cancer Med. 2021. 10: 416-23
22. Lüdemann W, Banan R, Hartmann C, Bertalanffy H, Di Rocco C. Pediatric intracranial primary anaplastic ganglioglioma. Childs Nerv Syst. 2017. 33: 227-31
23. Magrini S, Feletti A, Marton E, Longatti P. Gliomas of the pineal region. J Neurooncol. 2013. 115: 103-11
24. Mottolese C, Beuriat PA, Szathmari A. Pineal tumours: Experience of the French national register and the Lyon school, results and considerations. Neurochirurgie. 2015. 61: 223-35
25. Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2013-2017. Neuro Oncol. 2020. 22: 1-96
26. Packer RJ, Sutton LN, Rosenstock JG, Rorke LB, Bilaniuk LT, Zimmerman RA. Pineal region tumors of childhood. Pediatrics. 1984. 74: 97-102
27. Pagès A, Pagès M. Ganglioglioma of the pineal body. General review apropos of a case. Ann Anat Pathol (Paris). 1980. 25: 273-9
28. Pekmezci M, Villanueva-Meyer JE, Goode B, Van Ziffle J, Onodera C, Grenert JP. The genetic landscape of ganglioglioma. Acta Neuropathol Commun. 2018. 6: 47
29. Prabowo AS, van Thuijl HF, Scheinin I, Sie D, van Essen HF, Iyer AM. Landscape of chromosomal copy number aberrations in gangliogliomas and dysembryoplastic neuroepithelial tumours. Neuropathol Appl Neurobiol. 2015. 41: 743-55
30. Praver M, D’Amico R, Arraez C, Zacharia BE, Varma H, Goldman JE. Atypical pleomorphic neoplasms of the pineal gland: Case report and review of the literature. Surg Neurol Int. 2015. 6: 129
31. Radovanovic I, Dizdarevic K, de Tribolet N, Masic T, Muminagic S. Pineal region tumors-neurosurgical review. Med Arh. 2009. 63: 171-3
32. Rubinstein LJ, Okazaki H. Gangliogliomatous differentiation in a pineocytoma. J Pathol. 1970. 102: 27-32
33. Sajko T, Kudelić N, Lupret V, Lupret V, Nola IA. Treatment of pineal region lesions: Our experience in 39 patients. Coll Antropol. 2009. 33: 1259-63
34. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011. 121: 397-405
35. Schmincke A. Zur kenntnis der zirbelgeschwülste. Ein ganglioglioneurom der zirbel. Beitr Pathol Anat. 1930. 83: 279-88
36. Sireci AN, Aggarwal VS, Turk AT, Gindin T, Mansukhani MM, Hsiao SJ. Clinical genomic profiling of a diverse array of oncology specimens at a large academic cancer center: Identification of targetable variants and experience with reimbursement. J Mol Diagn. 2017. 19: 277-87
37. Tender GC, Smith RD. Pineal ganglioglioma in a young girl: A case report and review of the literature. J La State Med Soc. 2004. 156: 316-8
38. Tokoro K, Chiba Y, Ohtani T, Abe H, Yagishita S. Pineal ganglioglioma in a patient with familial basal ganglia calcification and elevated serum alpha-fetoprotein: Case report. Neurosurgery. 1993. 33: 506-11
39. 40. Woo PY, Teoh JY, Wong GK, Zhu XL, Siu DY, Kwan MC. A 45-year-old woman with reversible bilateral hearing loss. Neurology. 2013. 80: e23-6

