

- Department of Neurosurgery, University of Miami, Miami, Florida, United States.
- Department of Neurological Surgery, School of Medicine, Stanford University, Stanford, United States.
- Departments ofOtolaryngology, , Santa Clara Valley Medical Center, San Jose, California, United States.
- Departments of Pathology, Santa Clara Valley Medical Center, San Jose, California, United States.
- Departments of Neurosurgery, Santa Clara Valley Medical Center, San Jose, California, United States.
Correspondence Address:
Harminder Singh
Departments of Neurosurgery, Santa Clara Valley Medical Center, San Jose, California, United States.
DOI:10.25259/SNI_277_2020
Copyright: © 2020 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Annelise Sprau1, Anil Mahavadi1, Michael Zhang2, Micah Saste3, Michael Deftos4, Harminder Singh5. Rathke’s cleft cyst with xanthogranulomatous change: A case report and review of the literature. 15-Aug-2020;11:246
How to cite this URL: Annelise Sprau1, Anil Mahavadi1, Michael Zhang2, Micah Saste3, Michael Deftos4, Harminder Singh5. Rathke’s cleft cyst with xanthogranulomatous change: A case report and review of the literature. 15-Aug-2020;11:246. Available from: https://surgicalneurologyint.com/surgicalint-articles/10211/
Date of Submission
14-May-2020
Date of Acceptance
23-Jul-2020
Date of Web Publication
15-Aug-2020
Abstract
Background: Rathke’s cleft cysts (RCCs) are benign, typically asymptomatic sellar lesions found incidentally in adults, with a dramatically lower incidence in pediatric patients (
Case Description: The patient is a 17-year-old male with delayed puberty who presented with bitemporal hemianopsia and was found to have a 2.6 cm lesion, initially thought to be a craniopharyngioma. He subsequently underwent uncomplicated transsphenoidal endoscopic endonasal resection. Histology confirmed the diagnosis of RCC and demonstrated marked degenerative XGCs with squamous metaplasia. The patient tolerated the procedure well with improvement in visual symptoms.
Conclusion: RCC with XGC is a very rare pathology, particularly in the pediatric population. These lesions, while benign, can manifest clinically with significant symptoms. While treatment paradigms are not fully established with a small cohort of cases, endoscopic endonasal approaches have made surgical resection of these lesions a safe and effective treatment strategy, even in the pediatric population.
Keywords: Rathke’s cleft cyst, Skull base endoscopy, Xanthogranulomatous change
INTRODUCTION
Rathke’s cleft cysts (RCCs) are rare and benign sellar lesions that are derived from remnants of the Rathke’s pouch, an embryonic structure that gives rise to the anterior pituitary gland, pars intermedia, and pars tuberalis.[
In exceptional circumstances, RCCs can undergo xanthogranulomatous change (XGC), where leakage of cyst contents can lead to a granulomatous reaction characterized by cholesterol clefts, hemosiderin deposits, giant multinucleated cells, and inflammatory milieu along with infiltration of foamy or lipid-laden macrophages.[
CASE REPORT
A 17-year-old male presented to our neurosurgery clinic with progressive vision loss. He had initially been evaluated by a pediatric endocrinologist for delayed puberty as suggested by a high-pitched voice and feminine features. His hormonal work-up revealed panhypopituitarism. Preoperative Humphrey visual field testing demonstrated bitemporal visual field loss.
Further, magnetic resonance imaging (MRI) evaluation identified a 2.6 cm mass in the suprasellar region, noted to be hyperintense on T1 and T2 sequences with a nonenhancing, T1 hypointense nodule [
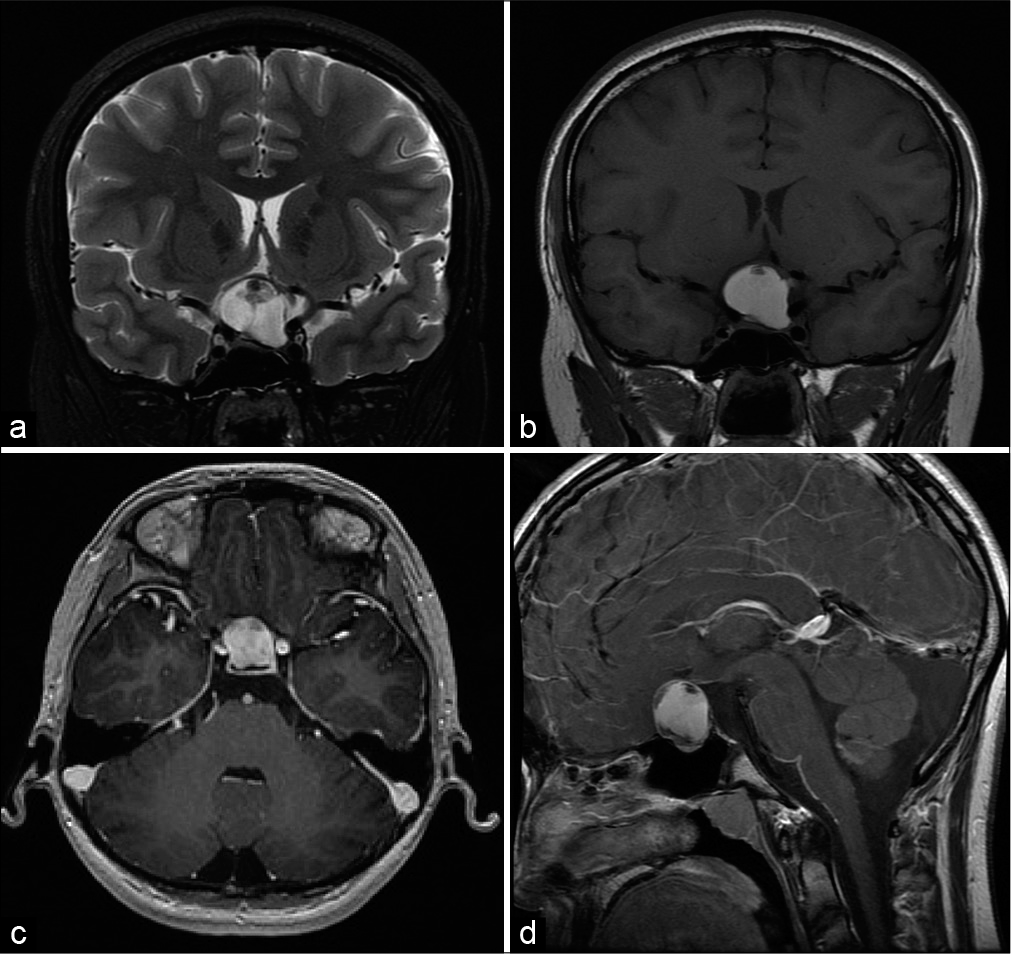
Figure 1:
Preoperative imaging (a) coronal T2-weighted magnetic resonance imaging (MRI) shows a hyperintense mass measuring 2.4 × 2.6 × 1.9 cm in the sellar and suprasellar region. (b) Coronal T1-weighted MRI shows a hyperintense mass with a hypointense, nonenhancing nodule. Postcontrast T1-weighted MRI in the axial (c) and sagittal (d) planes shows a heterogeneously enhancing mass.
Differential diagnosis included proteinaceous CP, cystic degeneration of a pituitary macroadenoma, and less likely RCC.
Given the vision changes, the patient and family consented to resection of the lesion through a transsphenoidal endoscopic endonasal approach. Since the presumptive diagnosis was that of a CP, we discussed with the patient that there was a distinct possibility that the pituitary stalk would have to be sacrificed to obtain gross total resection of the lesion.
The sella was accessed through the standard transsphenoidal approach with stereotactic navigation. After dural opening, the tumor was found to be firm and with a thick capsule. Sharp dissection and radiofrequency ablation were required to debulk the tumor. The tumor center yielded classic motor oil-like fluid as well as large cholesterol granulations that were histopathologically evaluated. There were dense adhesions of the tumor capsule to bilateral optic nerves and chiasm [
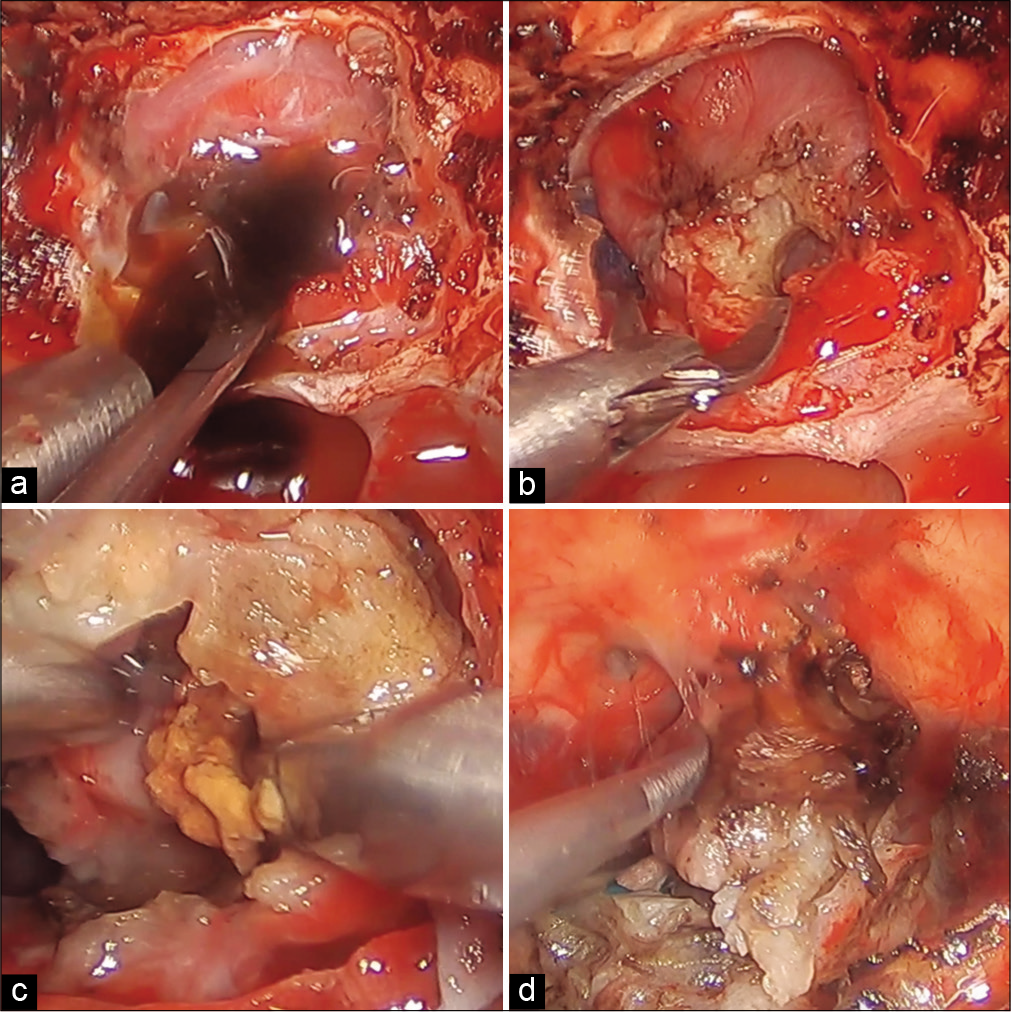
The capsule was sharply dissected away from the neural structures. We could now clearly visualize that the tumor capsule emanated from and had infiltrated the pituitary stalk. To achieve gross total resection, the stalk was truncated and the lesion was removed. Closure was performed using fat graft as inlay, followed by fascia lata wedged into the sellar opening using a MEDPOR (Stryker) graft, creating a gasket seal. This skull base reconstruction was then layered with a pedicled nasoseptal flap. The patient tolerated the procedure well.
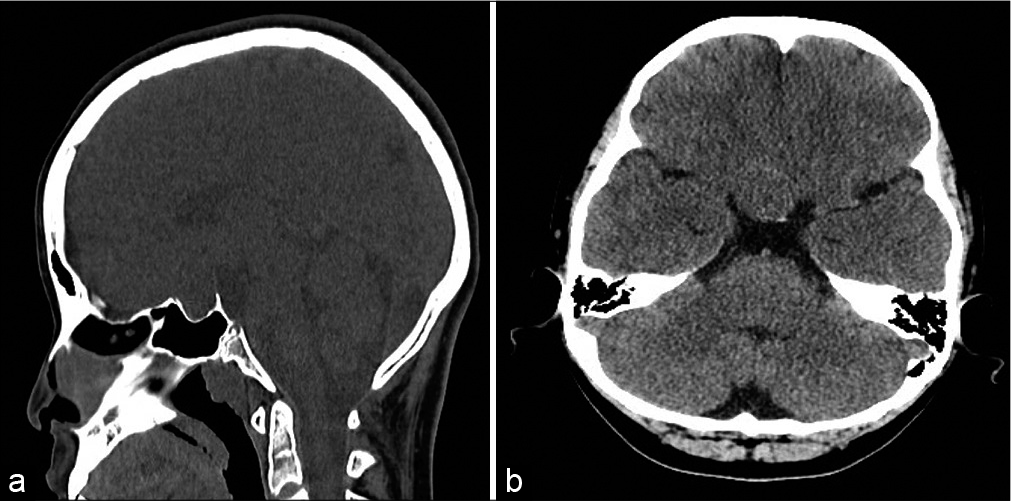
Postoperative Humphrey visual field testing demonstrated visual improvement after resection of the lesion and decompression of the chiasm. In addition to his preoperative hormone replacement regimen, he continued to require desmopressin acetate tablets at his 1-year postoperative follow-up. His postoperative 3-month MRI shows gross total resection of the lesion with decompression of the optic chiasm. The residual fat graft as well as the MEDPOR (Stryker) skull base reconstruction can also be visualized [
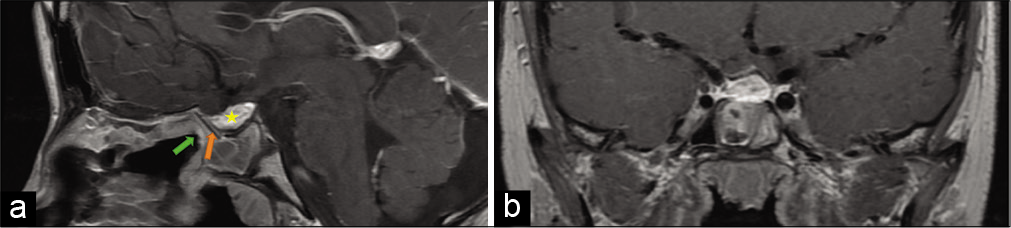
Figure 4:
Postoperative imaging postcontrast axial T1-weighted image in sagittal (a) and coronal (b) planes shows gross total resection of the tumor with decompression of the optic chiasm. The residual fat graft is noted in the inferior portion of the resection cavity (yellow star). The orange arrow in Figure 4a shows the placement of the Medpor graft, and the green arrow points to the layering of the pedicled nasoseptal flap for a multilayered closure.
Pathology showed a dense fibrous walled cyst with a focal residual internal epithelial lining consisting of squamous cells, whose membrane stained positive on beta-catenin stain [
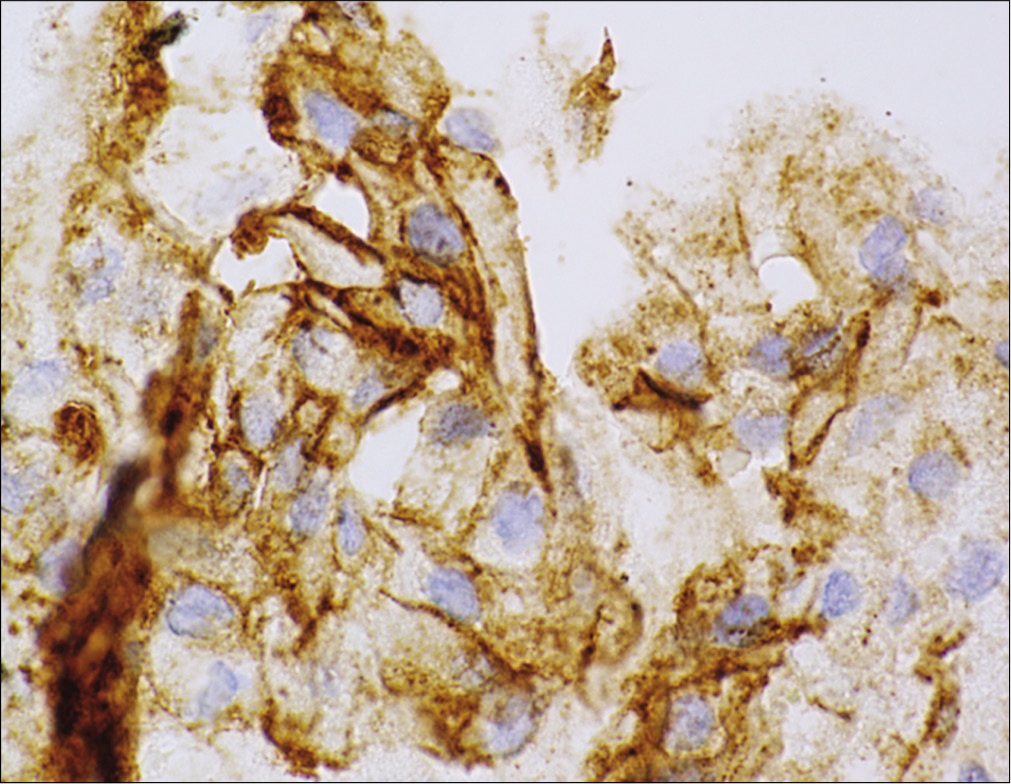
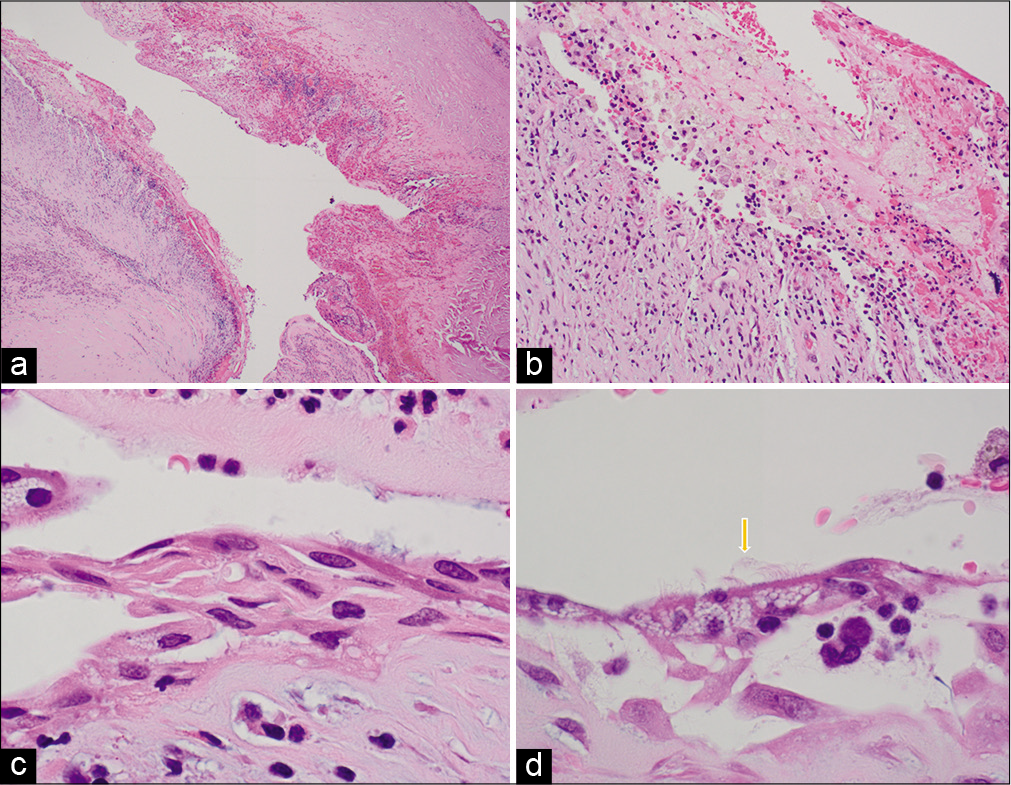
Figure 6:
Hematoxylin and eosin staining hematoxylin and eosin staining at ×4 (a), ×20 (b), ×100 (c), and ×1000 (d) demonstrating marked degenerative changes, lymphocytic/histiocytic inflammation, hemosiderin pigment, cholesterol clefts, and multinucleate giant cells. (d) Orange arrow shows the ciliated epithelial cells.
There were marked degenerative changes with lymphocytic and histiocytic inflammation, hemosiderin deposition, cholesterol clefts, and multinucleated giant cells. The histiocytes stained positive for CD68 and many contained a foamy cytoplasm that highlighted on periodic acid– Schiff diastase stain, which was consistent with them being muciphages. These features in summary were consistent with a RCC with marked XGC and squamous metaplasia.
DISCUSSION
We present the second reported occurrence of a suprasellar RCC with XGC in a pediatric patient [

Pathophysiology and presentation
Paulus et al. first postulated that XGCs were distinct entities, rather than degenerative changes of adamantinomatous craniopharyngiomas (ACs). In their retrospective analysis, they went on to report that patients with XGCs had histology consistent with massive cholesterol granuloma reactions, uncharacteristic of degenerative AC. In additional contrast, XGCs were predominantly associated with the second and third decades of life and with favorable outcomes.[
Thus, xanthogranuloma in the sellar region is suspected of being a terminal stage resulting from a secondary reaction caused by repeated inflammatory change, hemorrhage, and degeneration of a RCC.[
Clinical presentation is highly variable, as physical and neurological examination is dependent on time associated tolerance of the surrounding pituitary and optic anatomy. This is particularly challenging in pediatric populations given their fluctuating pubertal hormone levels.[
Medical and surgical management
Medical management of RCC with XGC while often can compensate for endocrinopathies may not adequately address the underlying cause and compression of neurovascular structures in the sellar region. Nakasu et al. outlined the medical management of a 23-year-old female with secondary amenorrhea using human chorionic gonadotropin-human menopausal gonadotropin. Although the patient’s secondary amenorrhea resolved as a result of the medical intervention, she continued to have persistently low luteinizing hormone levels. Further workup through MRI identified an RCC. The authors’ proceeded with surgical management, ultimately resolving this patient’s endocrinopathy.[
As evidenced by our own experience, as well as other reports in the literature, endoscopic skull base surgery has gained credence as an option to address midline anterior skull base pathology in the pediatric population.[
Because of clinically significant change in vision, our patient underwent endoscopic endonasal transsphenoidal approach for resection of this lesion. We achieved gross total resection without complications, and there was no recurrence or neurologic sequelae at 1-year follow-up. The other pediatric case in the literature was also treated endonasally and remained recurrence free at 6 months. It must be emphasized that because of the restrictive pediatric endonasal anatomy, as well as the nature of this pathology that leads to dense fibrous adhesions to neurovascular structures, endonasal resection should be limited to centers with multidisciplinary teams (neurosurgery, otolaryngology, endocrine, neuro-ophthalmology, and neuropathology) well versed in minimal access endoscopic techniques, as well as neurocritical care of the child.
The previous studies of the general pediatric RCC population have found 10-year survival rates to range from between 83% and 98% after complete resection. This drops to 20–70% over 10 years with recurrence.[
CONCLUSION
XGCs in RCCs are extremely rare granulomatous inflammatory reactions, particularly in the pediatric population. Clinical presentation will commonly affect vision and hormonal balance, inconsistently in children and teenagers. Surgical intervention remains an essential for tumor control, with the literature suggesting an excellent prognosis in cases where gross total resection can be achieved. Although the inflammatory components likely introduce fibrotic and adherent features, the lesion remains amenable to endoscopic endonasal resection in centers proficient in minimal access skull base endoscopy.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Albini CH, MacGillivray MH, Fisher JE, Voorhess ML, Klein DM. Triad of hypopituitarism, granulomatous hypophysitis, and ruptured Rathke’s cleft cyst. Neurosurgery. 1988. 22: 133-6
2. Amano K, Kubo O, Komori T, Tanaka M, Kawamata T, Hori T. Clinicopathological features of sellar region xanthogranuloma: Correlation with Rathke’s cleft cyst. Brain Tumor Pathol. 2013. 30: 233-41
3. Esiri M. Russell and Rubinstein’s pathology of tumors of the nervous system. Sixth edition. J Neurol Neurosurg Psychiatry. 2000. 68: 538
4. Gopal-Kothandapani JS, Bagga V, Wharton SB, Connolly DJ, Sinha S, Dimitri PJ. Xanthogranulomatous hypophysitis: A rare and often mistaken pituitary lesion. Endocrinol Diabetes Metab Case Rep. 2015. 2015: 140089
5. Kamoshima Y, Sawamura Y, Motegi H, Kubota K, Houkin K. Xanthogranuloma of the sellar region of children: Series of five cases and literature review. Neurol Med Chir (Tokyo). 2011. 51: 689-93
6. Karavitaki N, Wass JA. Non-adenomatous pituitary tumours. Best Pract Res Clin Endocrinol Metab. 2009. 23: 651-65
7. Kurisaka M, Fukui N, Sakamoto T, Mori K, Okada T, Sogabe K. A case of Rathke’s cleft cyst with apoplexy. Childs Nerv Syst. 1998. 14: 343-7
8. Li Z, Yang C, Bao X, Yao Y, Feng M, Deng K. Clinical features and treatment of secondary pituitary abscess after transsphenoidal surgery: A retrospective study of 23 Cases. World Neurosurg. 2018. 113: e138-45
9. Liu ZH, Tzaan WC, Wu YY, Chen HC. Sellar xanthogranuloma manifesting as obstructive hydrocephalus. J Clin Neurosci. 2008. 15: 929-33
10. Miyajima Y, Oka H, Utsuki S, Fujii K. Rathke’s cleft cyst with xanthogranulomatous change-case report. Neurol Med Chir (Tokyo). 2011. 51: 740-2
11. Mukherjee JJ, Islam N, Kaltsas G, Lowe DG, Charlesworth M, Afshar F. Clinical, radiological and pathological features of patients with Rathke’s cleft cysts: Tumors that may recur. J Clin Endocrinol Metab. 1997. 82: 2357-62
12. Muller HL, Gebhardt U, Faldum A, Warmuth-Metz M, Pietsch T, Pohl F. Xanthogranuloma, Rathke’s cyst, and childhood craniopharyngioma: Results of prospective multinational studies of children and adolescents with rare sellar malformations. J Clin Endocrinol Metab. 2012. 97: 3935-43
13. Nakasu Y, Nakasu S, Nakajima M, Itoh R, Matsuda M. Atypical Rathke’s cleft cyst associated with ossification. AJNR Am J Neuroradiol. 1999. 20: 1287-9
14. Nishimura F, Park YS, Motoyama Y, Nakagawa I, Yamada S, Tamura K. Intractable Rathke’s cleft cyst hidden behind co-existing giant pituitary adenoma-case report. World Neurosurg. 2019. 124: 9-11
15. Paulus W, Honegger J, Keyvani K, Fahlbusch R. Xanthogranuloma of the sellar region: A clinicopathological entity different from adamantinomatous craniopharyngioma. Acta Neuropathol. 1999. 97: 377-82
16. Raghunath A, Sampath S, Devi BI, Chandramouli BA, Lal GJ, Chickabasaviah YT. Is there a need to diagnose Rathke’s cleft cyst preoperatively?. Neurol India. 2010. 58: 69-73
17. Rahmani R, Sukumaran M, Donaldson AM, Akselrod O, Lavi E, Schwartz TH. Parasellar xanthogranulomas. J Neurosurg. 2015. 122: 812-7
18. Raper DM, Besser M. Clinical features, management and recurrence of symptomatic Rathke’s cleft cyst. J Clin Neurosci. 2009. 16: 385-9
19. Takanashi J, Tada H, Barkovich AJ, Saeki N, Kohno Y. Pituitary cysts in childhood evaluated by MR imaging. AJNR Am J Neuroradiol. 2005. 26: 2144-7
20. Tashiro T, Sano T, Xu B, Wakatsuki S, Kagawa N, Nishioka H. Spectrum of different types of hypophysitis: A clinicopathologic study of hypophysitis in 31 Cases. Endocr Pathol. 2002. 13: 183-95
21. Teramoto A, Hirakawa K, Sanno N, Osamura Y. Incidental pituitary lesions in 1, 000 unselected autopsy specimens. Radiology. 1994. 193: 161-4

