

- Department of Neurosurgery, University of Miami Miller School of Medicine, Miami, Florida, United States.
- Department of Neurological Surgery, University of Miami Miller School of Medicine, Miami, Florida, United States.
Correspondence Address:
Krisna Maddy, Department of Neurosurgery, University of Miami Miller School of Medicine, Miami, Florida, United States.
DOI:10.25259/SNI_616_2023
Copyright: © 2023 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Krisna Maddy1, Evan Luther1, Katherine Berry1, Victor M. Lu1, Ashish Shah1, Michael E. Ivan2, Ricardo J. Komotar1. Rathke’s cleft cysts causing Cushing’s disease: Two unique cases and review of the literature. 17-Nov-2023;14:402
How to cite this URL: Krisna Maddy1, Evan Luther1, Katherine Berry1, Victor M. Lu1, Ashish Shah1, Michael E. Ivan2, Ricardo J. Komotar1. Rathke’s cleft cysts causing Cushing’s disease: Two unique cases and review of the literature. 17-Nov-2023;14:402. Available from: https://surgicalneurologyint.com/surgicalint-articles/12638/
Date of Submission
23-Jul-2023
Date of Acceptance
19-Oct-2023
Date of Web Publication
17-Nov-2023
Abstract
Background: The presentation of isolated Rathke’s cleft cysts (RCC) without any associated pituitary adenoma in patients with symptoms consistent with Cushing’s disease (CD) remains exceedingly rare. As such, we aim to present two cases of RCC presenting with CD with a resultant resolution of their CD following surgical resection.
Case Description: Here, we present two cases of RCCs presenting with symptoms suggestive of CD. A functional pituitary microadenoma was the presumed diagnosis based on initial clinical presentation and diagnostic imaging suggesting a pituitary lesion. However, pathology results demonstrated no evidence of adenoma but cysts lined with columnar epithelia consistent with RCC. Complete surgical resection was achieved in both patients through endoscopic endonasal pituitary resection with postoperative symptomatic resolution and normalization of cortisol levels. In addition, we discuss the literature on this rare presentation and suggest a pathological mechanism for this unique presentation of RCC-causing CD.
Conclusion: Surgical resection of RCC may provide a “biochemical cure” for patients presenting with CD, as demonstrated by these two unique cases. The clinical features, histological findings, and possible pathological mechanisms for this unique presentation of RCC causing CD discussed lay the groundwork for future studies into the pathophysiology of RCC and CD.
Keywords: Cushing’s disease, Neurosurgery, Pituitary adenoma, Rathke’s cleft cyst
INTRODUCTION
Rathke’s cleft cysts (RCC) arise from the epithelial remnants of Rathke’s pouch, which persists in adult life between the pars anterior and pars posterior of the pituitary.[
The presence of RCC concurrently with pituitary adenoma is rare, however reported in the literature.[
CASE PRESENTATION #1
A 72-year-old male with a history of growth hormone deficiency symptomatic for generalized fatigue and weakness and on somatropin replacement therapy (1 mg daily) had an incidentally found pituitary lesion identified two decades prior on magnetic resonance imaging (MRI) following hospitalization for stroke. His comorbidities included obesity (body mass index, BMI 30) and diabetes. At that time, the lesion was assumed to be a microadenoma measuring 1.0 cm and abutting the optic chiasm. At the time, he had no visual deficits and was managed conservatively. In 2021, he presented to his outside endocrinologist with an elevated cortisol level of 2.3 mcg/dL from a normal value one year prior and the absence of overt changes in clinical features, suggesting subclinical CD. In addition, the low-dose (1 mg) dexamethasone suppression study did not suppress cortisol levels, indicating CD versus Cushing’s syndrome. Repeat imaging demonstrated the growth of the lesion, now measuring 1.3 cm, with no compression of the optic apparatus [
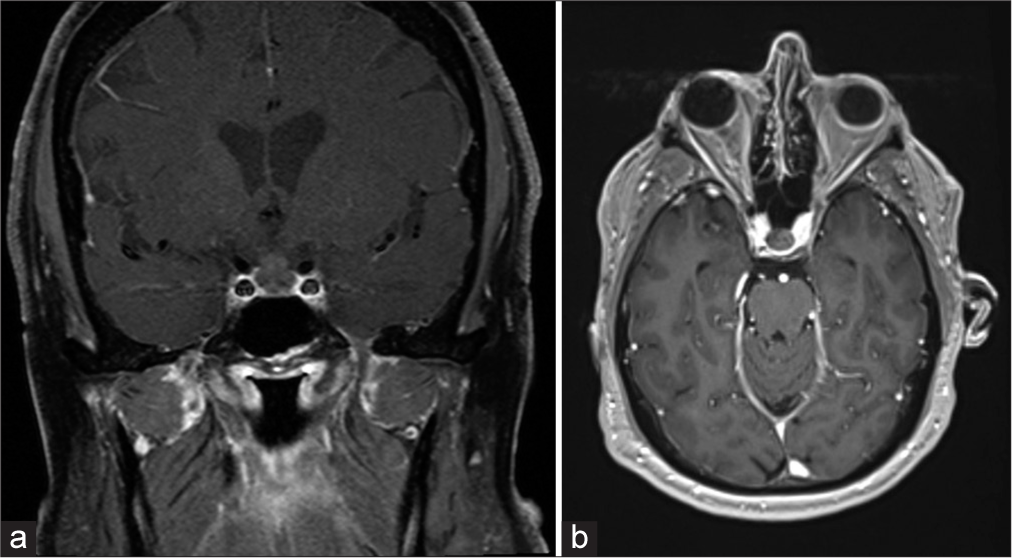
As a result of these findings, surgery was recommended. He underwent endoscopic endonasal transsphenoidal resection of the lesion without issue, and postoperative MRI demonstrated no residual mass [
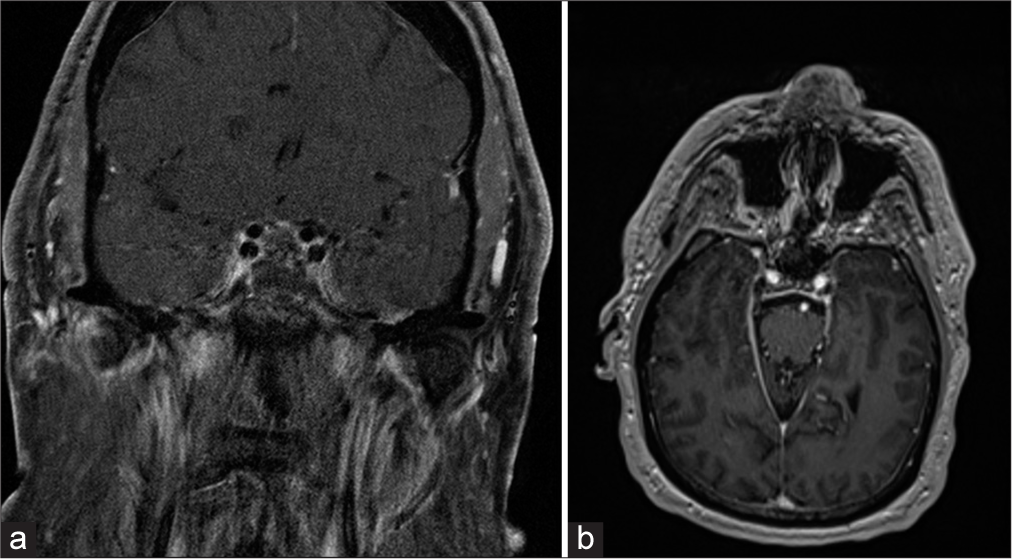
CASE PRESENTATION #2
A 61-year-old female initially presented in 2015 to her physician with a several-year history of symptoms of hirsutism, proximal muscle weakness, thinning skin, and easy bruising. Her comorbidities included obesity (BMI 37.6), diabetes, and hypertension. In June 2018, the patient’s serum ACTH was elevated at 77 pg/mL, and in February 2019, the patient’s urine-free cortisol was elevated at 134 mcg/dL. The patient reported a trial of mifepristone for several months but could not tolerate the medication. MRI in 2019 showed a 5 mm hypoenhancing lesion in the pituitary gland, presumed to be a pituitary microadenoma measuring approximately 3.5 mm [
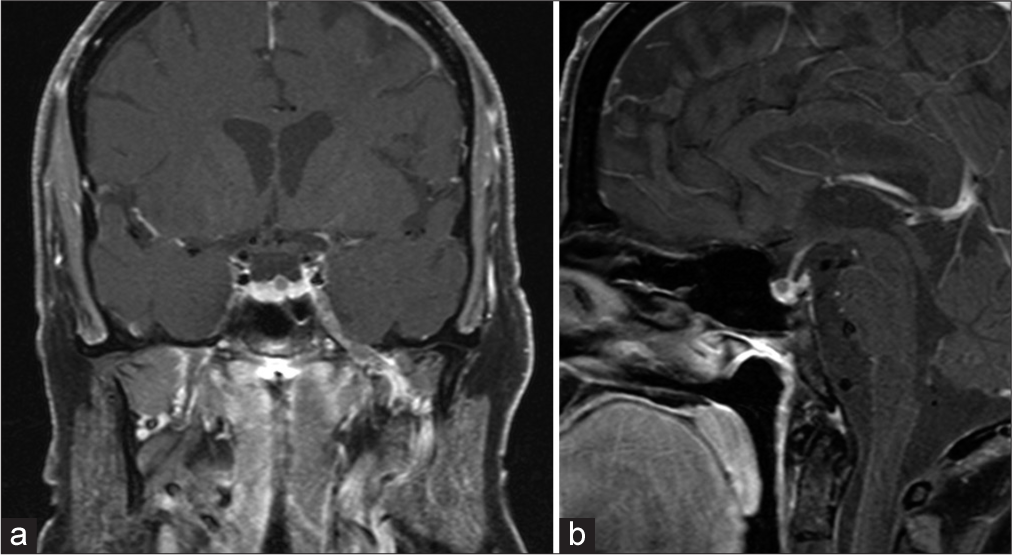
The patient underwent endoscopic endonasal transsphenoidal resection of the lesion, and postoperative MRI showed resection of the nodule in the mid-left superior adenohypophysis [
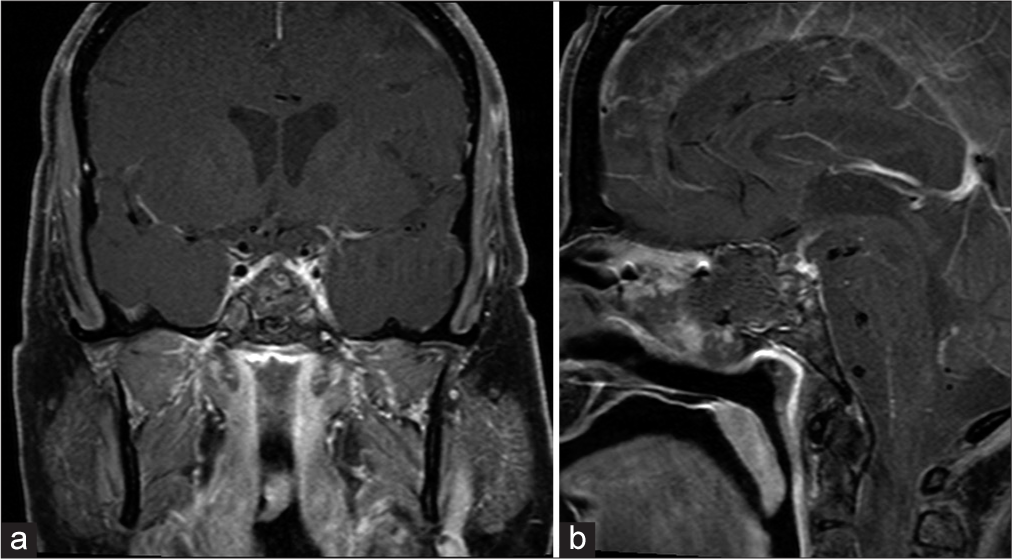
The pathology report of the specimen suggested RCC with Crooke’s hyaline changes. Noted on histology were several islands of eosinophilic acellular material, strips of ciliated epithelium with adjacent intact native pituitary parenchyma with all hormonal markers in a nonneoplastic distribution with no evidence of adenoma. The patient’s postoperative course was uneventful. One month postoperatively, the patient exhibited excellent recovery and no complications from the operation. On longer-term follow-up with endocrinology, the patient reported improvement in hirsutism and proximal muscle weakness.
DISCUSSION
To our knowledge, these are the first reported cases of RCC causing CD without concurrent pituitary adenoma or pituitary hyperplasia, as confirmed on histology. RCCs are considered embryological remnants of Rathke’s pouch, presenting typically as asymptomatic benign lesions.[
Typically, RCCs present on MRI as well-demarcated homogeneous lesions with a variable intensity highly dependent on cyst contents, ranging from clear cerebrospinal fluid-like fluid to thick mucoid material.[
RCCs are typically thought to not have functional disturbances compared to pituitary microadenomas; thus, the radiological correlation will suggest the latter’s presentation in a syndromic CD patient. Typically, an ACTH-producing adenoma would be suspected. The shared component of MRI presentation in both of our patients was the hypoenhanced/non-enhancing presentation of the lesion. Pituitary microadenomas also typically present as relatively hypointense lesions within an intensely enhanced pituitary gland.[
The histopathological intricacies in patients with other sellar lesions are also worth noting, as this is pivotal for accurate diagnosis, treatment planning, and prognostication, underscoring the significance of ongoing research efforts in this domain. Pituitary adenomas, the most prevalent sellar lesions, exhibit diverse histological patterns, as highlighted by the comprehensive classification by the World Health Organization, using markers of cytodifferentiation as the principal classifier.[
Understanding the histological presentation of RCCs may elucidate the pathophysiological mechanisms by which these lesions may have led to CD in our patients. CD typically occurs from a pituitary gland tumor or pituitary hyperplasia, resulting in the overproduction of ACTH and resultant overproduction of cortisol by the adrenal glands.[
Laboratory values reflecting endocrine dysfunction presented similarities and differences between these two cases [
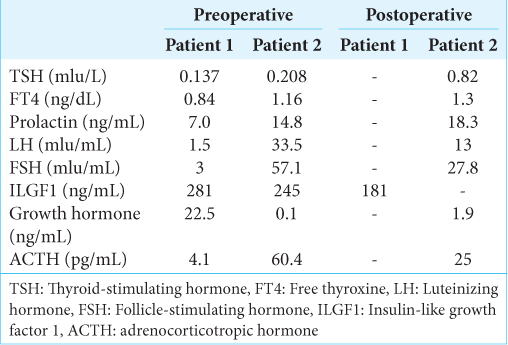
In measuring pre- and postoperative cortisol changes in both patients, we measured the degree of improvement that surgical resection of the RCC provided [

The surgical management of RCC is indicated in symptomatic presentations such as that in our patients. In previous studies of cases with overt signs of RCC compression causing symptomatic clinical presentation, there was demonstrated improvement in clinical symptoms after surgery.[
Suprasellar involvement of RCC can prove to be a neurosurgical challenge. Our first patient case presented with an abutment of the optic chiasm. It has been shown that compared with sellar-based RCCs, RCC’s visual dysfunction is more difficult to eliminate, recurs more frequently, and is associated with higher postoperative endocrine morbidity, and their preoperative visual dysfunction and headache less frequently improve with surgery.[
What remains to be understood in these cases is the pathophysiological mechanism by which these RCCs resulted in CD presentation, typically resulting from pituitary microadenomas. One possible explanation is that the RCC resulted in an overproduction of ACTH in neighboring cells surrounding the cyst. This suggests an effect of elevated circulating corticosteroids on the anterior pituitary parenchyma, resulting in the presentation of CD. In addition, the resolution of symptoms in both patients, including fatigue and generalized weakness, clinical and reduction of cortisol levels, and improvement in endocrinological laboratories postoperatively, suggests that the surgical removal of the RCC in these patients provided a “biochemical cure.”
However, the pathophysiologic mechanism of how the cystic cells resulted in symptomatic CD remains unclear. The absence of a concurrent pituitary microadenoma and benign pituitary tissue leaves no clear explanation of the cellular level changes that may have occurred. Nevertheless, the apparent symptomatic resolution of CD in these patients suggests that neurosurgical intervention was worthwhile. In cases of CD secondary to the presence of RCC, neurosurgical intervention should be considered for symptomatic improvement; however, further research should be undertaken to understand the underlying mechanism of this presentation.
CONCLUSION
Here, we present two cases of isolated RCCs presenting symptoms suggestive of CD. Based on the initial clinical presentation in these two patients and diagnostic imaging suggesting a pituitary lesion, pituitary microadenoma was the presumed diagnosis. However, there was no evidence of adenoma histopathologically, with findings more consistent with RCC. Maximal surgical resection was achieved in both patients through endoscopic endonasal pituitary resection with postoperative symptomatic resolution and normalization of cortisol levels. The clinical features, histological findings, and possible pathological mechanisms for this unique presentation of RCC-causing CD have been discussed and laid the groundwork for future studies into the pathophysiology of RCC and CD.
Declaration of patient consent
Patients’ consent not required as patients’ identities were not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The author confirms that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
1. Al-Brahim NY, Asa SL. My approach to pathology of the pituitary gland. J Clin Pathol. 2006. 59: 1245-53
2. Arita K, Uozumi T, Takechi A, Hirohata T, Pant B, Kubo K. A case of Cushing’s disease accompanied by Rathke’s cleft cyst: The usefulness of cavernous sinus sampling in the localization of microadenoma. Surg Neurol. 1994. 42: 112-6
3. Chaudhary V, Bano S. Imaging of the pituitary: Recent advances. Indian J Endocrinol Metab. 2011. 15: S216-23
4. Crenshaw WB, Chew FS. Rathke’s cleft cyst. AJR Am J Roentgenol. 1992. 158: 1312
5. Fairburn B, Larkin IM. A cyst of Rathke’s cleft. J Neurosurg. 1964. 21: 223-5
6. Jung JE, Jin J, Jung MK, Kwon A, Chae HW, Kim DH. Clinical manifestations of Rathke’s cleft cysts and their natural progression during 2 years in children and adolescents. Ann Pediatr Endocrinol Metab. 2017. 22: 164-9
7. Kageyama K, Oki Y, Nigawara T, Suda T, Daimon M. Pathophysiology and treatment of subclinical Cushing’s disease and pituitary silent corticotroph adenomas [Review]. Endocr J. 2014. 61: 941-8
8. Larkin S, Karavitaki N, Ansorge O. Rathke’s cleft cyst. Handb Clin Neurol. 2014. 124: 255-69
9. Laws ER, Lopes MB. The new WHO classification of pituitary tumors: Highlights and areas of controversy. Acta Neuropathol. 2006. 111: 80-1
10. Lonser RR, Nieman L, Oldfield EH. Cushing’s disease: Pathobiology, diagnosis, and management. J Neurosurg. 2017. 126: 404-17
11. Madhok R, Prevedello DM, Gardner P, Carrau RL, Snyderman CH, Kassam AB. Endoscopic endonasal resection of Rathke cleft cysts: Clinical outcomes and surgical nuances. J Neurosurg. 2010. 112: 1333-9
12. Miyagi A, Iwasaki M, Shibuya T, Kido G, Kushi H, Miyagami M. Pituitary adenoma combined with Rathke’s cleft cyst--case report. Neurol Med Chir (Tokyo). 1993. 33: 643-50
13. Nishio S, Mizuno J, Barrow DL, Takei Y, Tindall GT. Pituitary tumors composed of adenohypophysial adenoma and Rathke’s cleft cyst elements: A clinicopathological study. Neurosurgery. 1987. 21: 371-7
14. Oldfield EH, Vance ML, Louis RG, Pledger CL, Jane JA, Lopes MB. Crooke’s changes in cushing’s syndrome depends on degree of hypercortisolism and individual susceptibility. J Clin Endocrinol Metab. 2015. 100: 3165-71
15. Potts MB, Jahangiri A, Lamborn KR, Blevins LS, Kunwar S, Aghi MK. Suprasellar Rathke cleft cysts: Clinical presentation and treatment outcomes. Neurosurgery. 2011. 69: 1058-68
16. Raper DM, Besser M. Clinical features, management and recurrence of symptomatic Rathke’s cleft cyst. J Clin Neurosci. 2009. 16: 385-9
17. Sagan KP, Andrysiak-Mamos E, Sagan L, Nowacki P, Małkowski B, Syrenicz A. Cushing’s syndrome in a patient with Rathke’s cleft cyst and ACTH cell hyperplasia detected by (11)C-methionine PET imaging-a case presentation. Front Endocrinol (Lausanne). 2020. 11: 460
18. Sekine S, Shibata T, Kokubu A, Morishita Y, Noguchi M, Nakanishi Y. Craniopharyngiomas of adamantinomatous type harbor beta-catenin gene mutations. Am J Pathol. 2002. 161: 1997-2001
19. Shin JL, Asa SL, Woodhouse LJ, Smyth HS, Ezzat S. Cystic lesions of the pituitary: Clinicopathological features distinguishing craniopharyngioma, Rathke’s cleft cyst, and arachnoid cyst. J Clin Endocrinol Metab. 1999. 84: 3972-82
20. Stacchiotti S, Sommer J. Building a global consensus approach to chordoma: A position paper from the medical and patient community. Lancet Oncol. 2015. 16: e71-83
21. Wu W, Jia G, Jia W, Li G, Zhang J, Zhang L. Pituitary adenoma associated with Rathke’s cleft cyst: Report of 15 cases. Can J Neurol Sci. 2018. 45: 68-75

