

- Department of Neurosurgery, Fukuoka Children’s Hospital, Fukuoka, Japan
- Department of Neurosurgery, Kyushu University, Fukuoka, Japan
- Department of Psychiatry, Shourai Hospital, Fukuoka, Japan
- Department of Orthopedic Surgery, Fukuoka Children’s Hospital, Fukuoka, Japan
- Department of Neurosurgery, Harasanshin Hospital, Fukuoka, Japan.
Correspondence Address:
Ai Kurogi, Department of Neurosurgery, Fukuoka Children’s Hospital, Fukuoka, Japan.
DOI:10.25259/SNI_156_2023
Copyright: © 2023 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Ai Kurogi1, Nobuya Murakami1, Takafumi Shimogawa2, Nobutaka Mukae2, Satoshi O. Suzuki3, Toru Yamaguchi4, Koji Yoshimoto2, Takato Morioka5. Severe type of segmental spinal dysgenesis with complete disconnection of the spinal cord and vertebra associated with open neural tube defect. 28-Apr-2023;14:149
How to cite this URL: Ai Kurogi1, Nobuya Murakami1, Takafumi Shimogawa2, Nobutaka Mukae2, Satoshi O. Suzuki3, Toru Yamaguchi4, Koji Yoshimoto2, Takato Morioka5. Severe type of segmental spinal dysgenesis with complete disconnection of the spinal cord and vertebra associated with open neural tube defect. 28-Apr-2023;14:149. Available from: https://surgicalneurologyint.com/surgicalint-articles/12303/
Date of Submission
14-Feb-2023
Date of Acceptance
05-Apr-2023
Date of Web Publication
28-Apr-2023
Abstract
Background: Severe type of segmental spinal dysgenesis (SSD) is a rare and complex anomaly in which the spinal cord completely disconnects at the portion of the spinal dysgenesis. Although closed spinal dysraphisms have been associated with SSD, to the best of our knowledge, the association between open neural tube defect (ONTD) and SSD is significantly rare, with only one case being reported to date.
Case Description: We report a case of an infant with severe SSD and a disconnected spinal cord and spinal column at the thoracolumbar junction associated with myelomeningocele (MMC) in the lumbosacral region. The patient presented severe neurological deficits in the legs and impaired bowel function. The spinal column of L1–L3 was absent. The lower spinal segment consisted of neural placode at the L5–S1 level and no connecting structure between the upper and lower spinal cords. A repair surgery for MMC, including cord untethering and dura plasty, was performed. Histopathological findings revealed a neural placode consisting of a neuroglial tissue and leptomeninges.
Conclusion: The management of severe SSD during the perinatal period is more challenging when it is associated with ONTD. We report detailed neuroradiological, intraoperative, and histological findings of such a case and discuss the embryopathogenesis of the associated ONTD and the treatment strategies.
Keywords: Gastrulation, Junctional neural tube defect, Neurulation failure, Open neural tube defect, Segmental spinal dysgenesis
INTRODUCTION
Segmental spinal dysgenesis (SSD) is a rare congenital abnormality in which a segment of the spine and spinal cord fails to develop appropriately, and in most severe cases, the spinal cord completely disconnects at the portion of the spinal dysgenesis, presenting with severe neurological deficits.[
Herein, we present detailed neuroradiological, intraoperative, and histological findings of the case of an infant with severe SSD and a disconnected spinal cord and spinal column at the thoracolumbar junction associated with MMC in the lumbosacral region.
CASE REPORT
A 34-year-old primigravida woman was referred to our hospital as no movements were detected in the fetus’s legs on transabdominal ultrasound examination at 26 + 5 weeks of gestation. Prenatal MRI performed at 27 + 4 weeks of gestation failed to reveal apparent abnormalities in the lumbosacral regions, hydrocephalus, or a Chiari malformation.
The mother vaginally delivered a girl weighing 2,471 g and with an Apgar score of 8 at 37 + 4 weeks of gestation. A skin defect (15 × 25 mm) associated with a dysplastic neural placode exposed to the surface was detected in the infant at the lumbosacral region without cerebrospinal fluid (CSF) leakage [
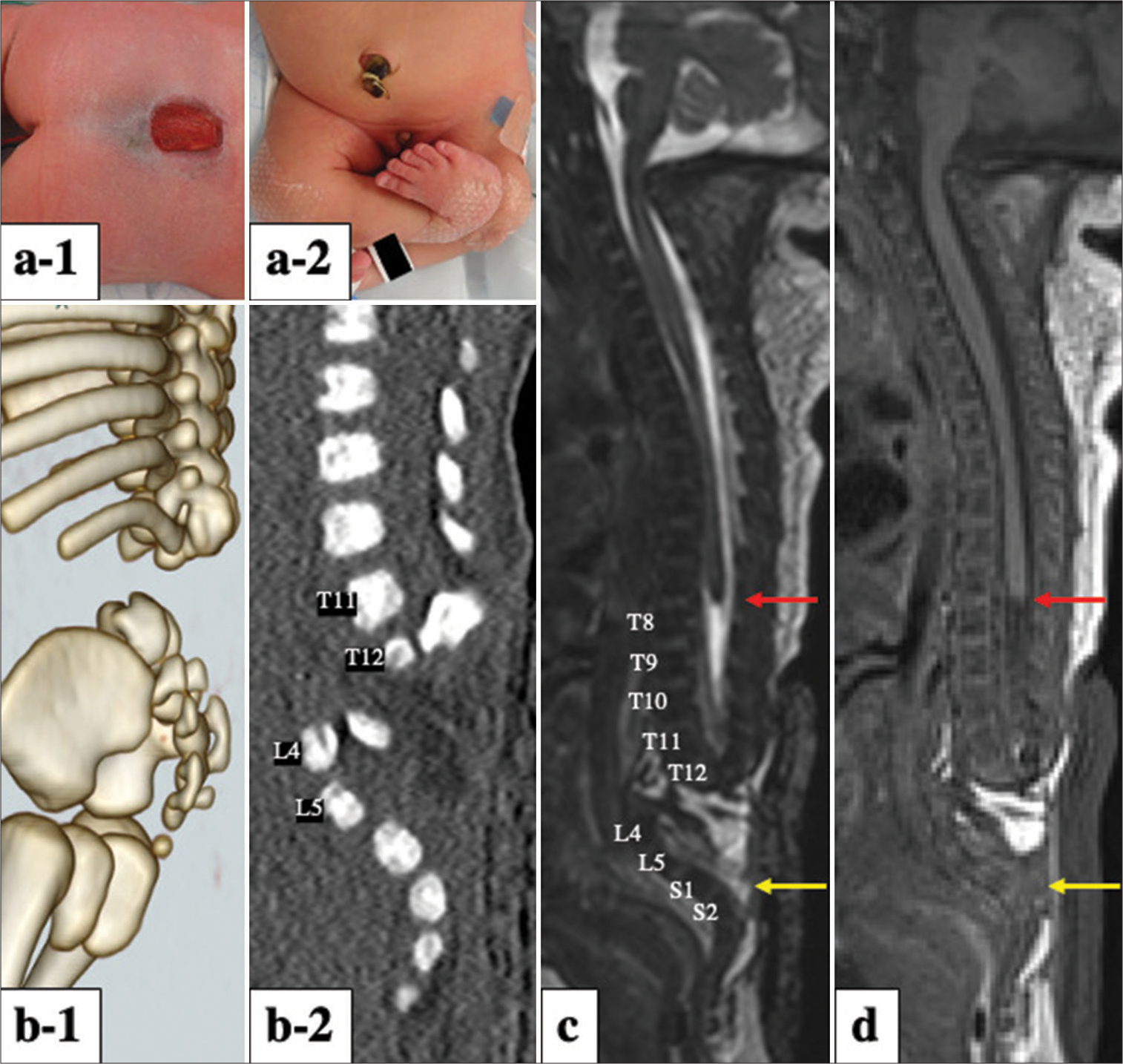
Figure 1:
Photographs at birth showing (a-1) a neural placode at the lumbosacral region with an open skin defect (15 × 25 mm) exposed to the surface and (a-2) the patient’s lower limbs in the cross-legged position with severe flexor contractures. (b-1) Three-dimensional and (b-2) sagittal spinal computed tomography images showing a complete absence of the L1–L3 lumbar vertebrae and dislocation of the lower segment, which deviates anteriorly at the level of the absence. (c) Sagittal three-dimensional-heavily T2-weighted and (d) sagittal T1-weighted spoiled gradient-recalled echo images also demonstrated two completely separated spinal cord segments. The upper segment ends at T8, with a “cigar-shaped” appearance instead of the usual tapered conus (red arrow). The lower segment is located from L4 to S1, displaying an exposed placode (yellow arrow) lying flush with the skin surface with an underlying subarachnoid space, consistent with myelomeningocele.
Spinal three-dimensional (3D) computed tomography revealed that the spinal column at L1–L3, including both vertebrae and posterior elements, was absent and that the lumbar spine below L4 and bifid sacrum deviated anteriorly [
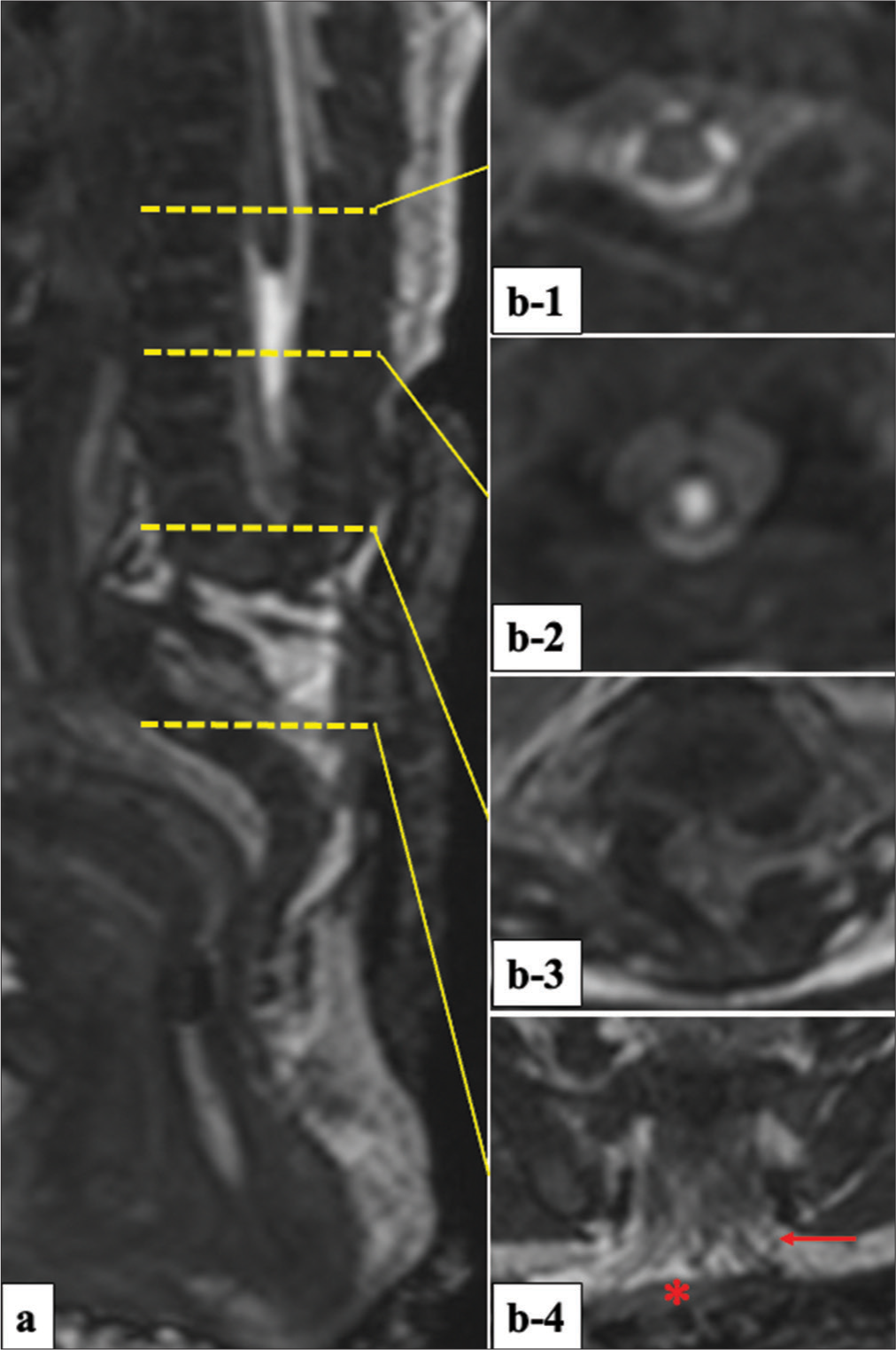
Figure 2:
A sagittal view (a) and serial axial views (b-1-4) of 3D-hT2WI delineating detailed structures. (b-1) Normal appearance of upper spinal cord at T7 level. (b-2) Absence of band-like structure caudally to the upper spinal cord. (b-3) No signal intensity of CSF at the portion between the upper and lower dural sac. (b-4) Bundle of nerve roots (red arrow) in the subarachnoid space and the neural placode (red asterisk) attached to the skin surface. 3D-hT2WI: Three-dimensional heavily T2-weighted imaging, CSF: Cerebrospinal fluid.
On the day after birth, the infant underwent repair surgery for MMC. The open neural placode was circumferentially resected from the surrounding skin, and proximal spinal cord and nerve roots continuing with the ventral side of the neural placode were observed in the open dural sac filled with CSF [
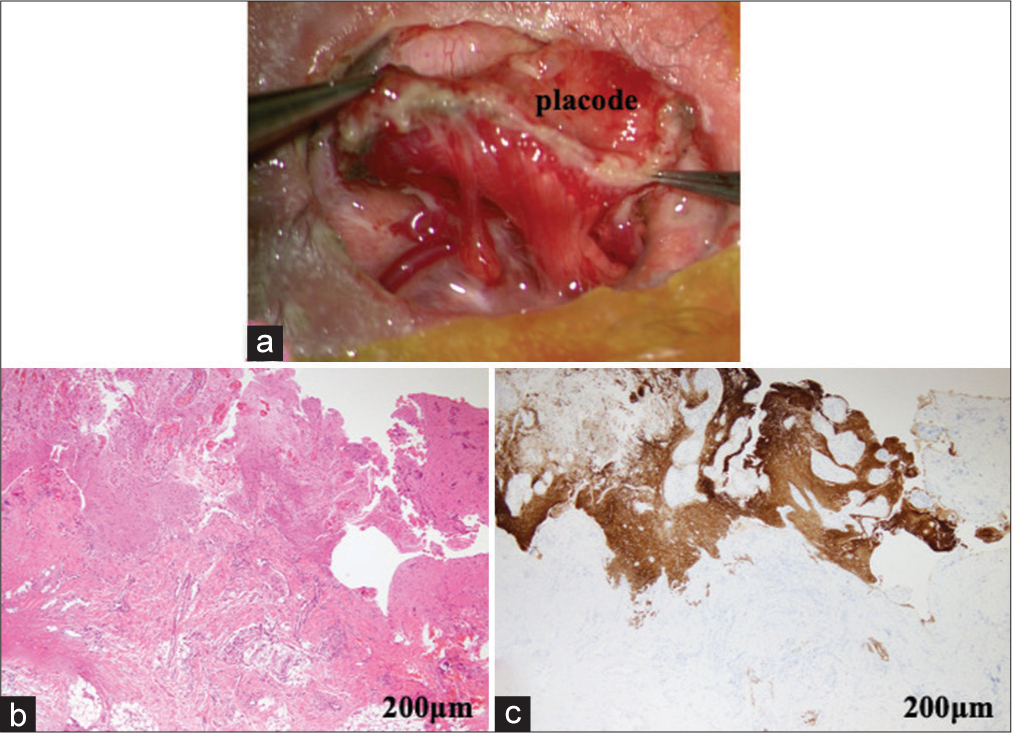
Figure 3:
(a) An intraoperative picture of the neural placode resected from the attached skin, demonstrating proximal spinal cord and nerve roots arising from the dorsal aspect of the placode. Histopathological findings of the resected margin of the neural placode stained with hematoxylin and eosin (b) and immunostained for glial fibrillary acidic proteins (GFAP, c). The specimen comprises a GFAP-immunopositive neuroglial tissue including mature neurons and an adjoining fibrotic leptomeningeal tissue.
The histopathological examination of the resected tissue from the margin of the neural placode showed a glial fibrillary acidic protein-immunopositive neuroglial tissue adjoining a fibrotic leptomeningeal tissue [
DISCUSSION
The embryopathogenesis of SSD has been hypothesized to be the positioning error of the chordomesoderm cells, which is the mesoderm that gives rise to the notochord. The error causes apoptosis of the cells in the malpositioned segment (positional apoptosis) during gastrulation.[
Although rare coexistence of ONTD in SSD cases has been explained by the findings of studies on avian embryos demonstrating that removal of the notochordal segment does not affect the formation of dermatome and myotome (future skin and muscle, respectively) but causes defects of sclerotome,[
The morphological feature of SSD in the aspect of the disconnected upper and lower spinal cords has been advocated to be similar to that of a new disease entity, called a junctional neural tube defect (JNTD).[
Considering that most cases of SSD have severe bladder dysfunction as they age,[
In the present case, we performed dura plasty for the MMC immediately after birth to prevent infection. Regarding cord tethering, severe cases of SSD were unlikely to benefit from untethering surgery because the neurological disturbances appeared to be related to congenital hypoplasia of the nerve roots and spinal cord, and none of the studies have reported progressive neurological deficits during the follow-up periods.[
Patients with SSD require orthopedic treatment to establish and maintain spinal stability and to slow the progressive worsening of neurological functioning due to kyphosis.[
CONCLUSION
Severe SSD has a complex and serious abnormality and earlier neurosurgical intervention is required during the perinatal period in cases associated with ONTD. In the future, studies including a large number of patients are warranted for further understanding of this congenital disorder.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Research Foundation of Fukuoka Children’s Hospital.
Conflicts of interest
There are no conflicts of interest.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
Acknowledgment
We would like to thank Drs. Tadamune Kinjyo and Naoyuki Nakanami, Departments of Neonatology and Obstetrics, respectively, for supporting our study.
References
1. Ali M, McNeely PD. Junctional neural tube defect: A supporting case report. Childs Nerv Syst. 2018. 34: 1447-8
2. Bristol RE, Theodore N, Rekate HL. Segmental spinal dysgenesis: Report of four cases and proposed management strategy. Childs Nerv Syst. 2007. 23: 359-64
3. Chellathurai A, Ayyamperumal B, Thirumaran R, Kathirvelu G, Muthaiyan P, Kannappan S. Segmental spinal dysgenesis-“redefined. ” Asian Spine J. 2019. 13: 189-97
4. Dady A, Havis E, Escriou V, Catala M, Duband JL. Junctional neurulation: A unique developmental program shaping a discrete region of the spinal cord highly susceptible to neural tube defects. J Neurosci. 2014. 34: 13208-21
5. De Groat WC, Griffiths D, Yoshimura N. Neural control of the lower urinary tract. Compr Physiol. 2015. 5: 327-96
6. Dias MS, Walker ML. The embryogenesis of complex dysraphic malformations: A disorder of gastrulation?. Pediatr Neurosurg. 1992. 18: 229-53
7. Eibach S, Moes G, Hou YJ, Zovickian J, Pang D. Unjoined primary and secondary neural tubes: Junctional neural tube defect, a new form of spinal dysraphism caused by disturbance of junctional neurulation. Childs Nerv Syst. 2017. 33: 1633-47
8. Eibach S, Pang D. Junctional neural tube defect. J Korean Neurosurg Soc. 2020. 63: 327-37
9. Faciszewski T, Winter RB, Lonstein JE, Sane S, Erickson D. Segmental spinal dysgenesis: A disorder different from spinal agenesis. J Bone Joint Surg. 1995. 77: 530-7
10. Florea SM, Faure A, Brunel H, Girard N, Scavarda D. A case of junctional neural tube defect associated with a lipoma of the filum terminale: A new subtype of junctional neural tube defect?. J Neurosurg Pediatr. 2018. 21: 601-5
11. Hashiguchi K, Morioka T, Murakami N, Yamashita K, Hiwatashi A, Ochiai M. Clinical significance of prenatal and postnatal heavily T2-weighted magnetic resonance images in patients with myelomeningocele. Pediatr Neurosurg. 2015. 50: 310-20
12. Hashiguchi K, Morioka T, Yoshida F, Miyagi Y, Mihara F, Yoshiura T. Feasibility and limitation of constructive interference in steady-state (CISS) MR imaging in neonates with lumbosacral myeloschisis. Neuroradiology. 2007. 49: 579-85
13. Hirano S, Hirako R, Kajita N, Norita M. Morphological analysis of the role of the neural tube and notochord in the development of somites. Anat Embryol (Berl). 1995. 192: 445-57
14. Kaplan KM, Spivak JM, Bendo JA. Embryology of the spine and associated congenital abnormalities. Spine J. 2005. 5: 564-76
15. Kashgari AA. Segmental spinal dysgenesis with open spinal dysraphism and Chiari II features, case report. Radiol Case Rep. 2020. 15: 1965-7
16. Marín-Padilla M. Cephalic axial skeletal-neural dysraphic disorders: Embryology and pathology. Can J Neurol Sci. 1991. 18: 153-69
17. Moore K, Persaud TV, Torchia M, editors. The Developing Human: Clinically Oriented Embryology. Philadelphia, PA: Saunders; 2018. p.
18. Morioka T, Hashiguchi K, Mukae N, Sayama T, Sasaki T. Neurosurgical management of patients with lumbosacral myeloschisis. Neurol Med Chir. 2010. 50: 870-6
19. Pavlova OM, Ryabykh SO, Kozyrev DA, Gubin AV. Surgical treatment of thoracolumbar segmental spinal dysgenesis: Optimal type of fusion. World Neurosurg. 2017. 106: 551-6
20. Schmidt C, Voin V, Iwanaga J, Alonso F, Oskouian RJ, Topale N. Junctional neural tube defect in a newborn: Report of a fourth case. Childs Nerv Syst. 2017. 33: 873-5
21. Scott RM, Wolpert SM, Bartoshesky LE, Zimbler S, Karlin L. Segmental spinal dysgenesis. Neurosurgery. 1988. 22: 739-44
22. Tortori-Donati P, Fondelli MP, Rossi A, Raybaud CA, Cama A, Capra V. Segmental spinal dysgenesis: Neuroradiologic findings with clinical and embryologic correlation. Am J Neuroradiol. 1999. 20: 445-56
23. Tortori-Donati P, Rossi A, Cama A. Spinal dysraphism: A review of neuroradiological features with embryological correlations and proposal for a new classification. Neuroradiology. 2000. 42: 471-91
24. Wang KC, Lee JS, Kim K, Im YJ, Park K, Kim KH. Do junctional neural tube defect and segmental spinal dysgenesis have the same pathoembryological background?. Childs Nerv Syst. 2020. 36: 241-50

