

- Department of Neurology, Complexo Hospital de Clínicas da UFPR, Curitiba, Paraná, Brazil.
DOI:10.25259/SNI_861_2020
Copyright: © 2020 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Otto Hernandez Fustes, Cláudia Suemi Kamoi Kay, Paulo José Lorenzoni, Renata Dal-Prá Ducci, Lineu Cesar Werneck, Rosana Herminia Scola. Somatosensory evoked potentials in Hirayama disease: A Brazilian study. 22-Dec-2020;11:464
How to cite this URL: Otto Hernandez Fustes, Cláudia Suemi Kamoi Kay, Paulo José Lorenzoni, Renata Dal-Prá Ducci, Lineu Cesar Werneck, Rosana Herminia Scola. Somatosensory evoked potentials in Hirayama disease: A Brazilian study. 22-Dec-2020;11:464. Available from: https://surgicalneurologyint.com/surgicalint-articles/10489/
Date of Submission
01-Dec-2020
Date of Acceptance
04-Dec-2020
Date of Web Publication
22-Dec-2020
Abstract
Background: Hirayama’s disease (HD) is characterized by an insidious onset asymmetric weakness and atrophy of the forearm and hand. Taking as a premise, the etiopathogenesis of the disease is attributed to forward displacement of posterior wall of lower cervical dural canal in neck flexion causing marked compression and flattening of lower spinal cord. This may result in compression of the posterior column of the spinal cord and seems likely to result in somatosensory evoked potentials (SSEPs) abnormalities. In the present study, we studied the possible involvement of the lemniscal dorsal pathway in patients with HD.
Methods: SSEPs in upper and lower extremities were prospectively performed in eight patients with HD. All the patients were recruited from the outpatient clinic of a neuromuscular disorder center from South Brazil. SSEPs were obtained by transcutaneous electrical stimulation of the median and posterior tibial nerves, on both sides. We collected the amplitude and the latency of the different components obtained in each channel. The interpretation was based on Brazilian study standards.
Results: We evaluated seven men and one woman (mean age 27). The data obtained were compared to a control group consisting of eight patients with spondylotic cervical myelopathy, 6 men and 2 women with mean age of 59 years. The measurements of obtained by the SSEP were also compared between the groups and no significant difference was found for any of them.
Conclusion: SSEP did not turn out to be an electrophysiological marker in our HD patients.
Keywords: Cervical spondylopathy, Hirayama disease, Monomelic amyotrophy, Somatosensory evoked potentials
INTRODUCTION
Hirayama’s disease (HD), also known as juvenile muscular atrophy of distal upper extremity, was first described by Keizo Hirayama in 1959.[
HD is a benign, self-limiting pathology; after a progressive phase of the neurological deficits affecting the C7, C8, and T1 myotomes for about 1–5 years, it has a spontaneous arrest. The first autopsy from a patient died of lung carcinoma, was published in 1987, and revealed decreased number of both large and small nerve cells and degenerative changes, anteroposterior flattening, and ischemic changes in the anterior horn cells of the lower cervical cord segment and suggested circulatory insufficiency in the lower cervical cord as the leading cause.[
Atopy and elevated serum IgE level has also been postulated to be precipitating factors in HD.[
The differential diagnosis of HD includes the distal form of spinal muscular atrophy, amyotrophic lateral sclerosis (ALS), postpolio syndrome, multifocal motor neuropathy with conduction block, and toxic neuropathy as well as structural lesions of the cervical cord. Their typical clinical, radiological, and electrophysiological features can identify these clinical entities.
Taking as a premise, the etiopathogenesis of the disease is attributed to forward displacement of posterior wall of lower cervical dural canal in neck flexion causing marked compression and flattening of lower spinal cord. This may result in compression of the posterior column of the spinal cord and seems likely to result in somatosensory evoked potentials (SSEPs) abnormalities. The aim of this study was to evaluate the somatosensory pathways through studies of SSEP, to evaluate conduction between the dorsal column of the spinal cord dorsal horn and the sensory cortex.
In the first phase of the project, theoretical reflective research was carried out, which combined the deductive method with the bibliographic research technique, based on articles published in English or Portuguese in the database of PubMed and Lilacs. The terms that were used included “Somatosensory evoked potentials AND Hirayama disease;” “evoked potential AND Hirayama disease;” “Hirayama disease AND neurophysiology;” “Hirayama disease AND Latin American;” and “somatosensory evoked potentials AND Monomelic amyotrophic.”
MATERIALS AND METHODS
The SSEPs in upper and lower extremities were prospectively performed in eight patients with HD. All the patients born and living in South Brazil, were recruited from the outpatient clinic of a neuromuscular disorder center at the Hospital de Clínicas of the Federal University of Paraná, Curitiba, Brazil.
All of our patients met the diagnostic criteria for HD including (a) onset before the age of 25 years; (b) unilateral or bilateral distal predominant weakness and wasting of the upper limbs without sensory impairment; (c) static clinical course after slowly insidious progression; (d) no lower limb involvement; and (e) no history of syringomyelia, spinal cord tumor, cervical vertebral abnormality, multifocal motor neuropathy, congenital muscular dystrophy, trauma, inflammation, infection, or any other cause for the clinical findings.[
SSEPs were obtained by transcutaneous electrical stimulation of the median nerve at the wrist and the posterior tibial nerve at the ankle, on both sides, and were alternately recorded as recommended by the International Federation of Clinical Neurophysiology[
Potentials were identified on the curve obtained by averaging 2 series of 500 acquisitions reproducible for each stimulated nerve with the target muscle minimally active. We collected the amplitude and the latency of the different components obtained in each channel. The latency was measured at the peak amplitude, and the amplitude was measured between the peak and the end of the potential. The interpretation was based on our laboratory standards using the same technique and apparatus.
Data from two groups of patients were provided, the group with HD being called “case” and the group with cervical spondylopathy called “control.” The groups were compared regarding age, sex, the result of studying the SSEP (N9, N20, N21, and P40), and the presence of altered measures. The components examined were absolute latencies and interpeaks of N9 and N20 in the upper limbs and absolute latencies and interpeaks of N21 and P40 in the lower limbs.
To compare proportions, Fisher’s exact test was used, and to compare quantitative measures, the Mann–Whitney U-test was used. Quantitative measurements were represented by the median and interquartile interval (first quartile; third quartile).
The study was approved by the Local Ethics Committee for Human Research of the Hospital de Cliínicas da Universidade Federal do Paraná, with a number 4.340.521. All studies were conducted in accordance with ethical principles after obtaining patient informed consent.
RESULTS
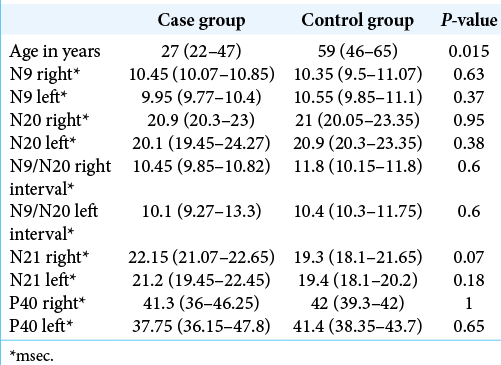
We evaluated seven men and one woman (mean age 27). The data obtained were compared to a control group consisting of eight patients with spondylotic cervical myelopathy, six men and two women with mean age of 59 years old.
Comparison of proportions
No difference was found between the proportions of gender or alterations in the SSEP between the groups. The comparison between the proportions of changes was made considering both the right and left sides separately and together [
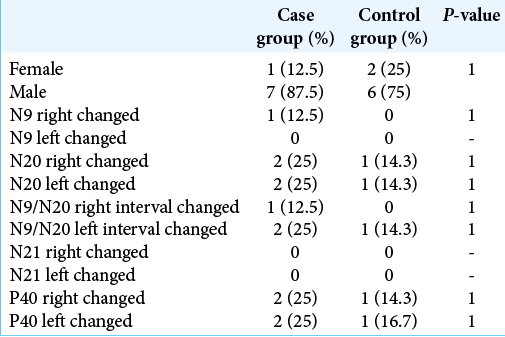
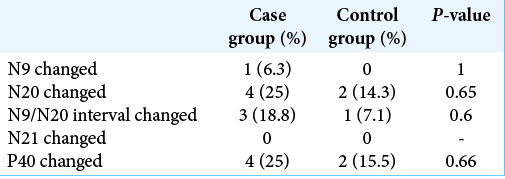
Comparison of measures
A significant difference was found between the ages of the groups, with the case group having a median age below that of the control group.
The measurements obtained by the SSEP were also compared between the groups and no significant difference was found for any of them.
In three patients, we performed the SSEP study with neck flexion, which showed no differences in relation to the baseline examination.
Asymmetry
The proportion of altered results was compared within the group of cases between the right and left sides, but there was no significant difference.
DISCUSSION
From the best of our knowledge, in the literature review carried out (based on Scielo and PubMed websites), we did not find a similar study in Latin America. Only case reports or series with image studies.[
HD is considered to be a benign focal motor neuron disease, muscle weakness and wasting is most pronounced in distal muscles of the upper limb, nerve conduction studies are not required to make the diagnosis, but may be used to exclude other conditions. The electromyography results in Hirayama syndrome show denervation of atrophied muscles and in over 90% of patients also show contralateral denervation of the same muscles.[
The motor nerve conduction velocities are normal, except occasional minimal slowing in the ulnar nerve. The compound muscle action potentials have reduced amplitudes in the atrophied muscles.[
On the other hand, although there have been significant advances in the HD image, the characteristic findings of studies of SSEP in HD patients remain controversial, due to the discrepancies of the findings in the few studies. Restuccia et al. in a study of upper limb SSEPs demonstrated that neck flexion caused a significant amplitude decrease of the N13 cervical response in patients with HD but not in patients with ALS and healthy subjects, suggesting that direct cord compression or microvascular changes could in theory account for this position-related dysfunction.[
However, Misra et al. found no significant change in SSEPs and F-wave parameters on neck flexion compared with neutral position in their study of eight patients of HD and controls.[
We did not find another study in the literature that evaluated the P40 potential. In our patients, there was no significant impairment of the gracile fasciculus.
The key to diagnose this disease is based on the typical clinical features and dynamic MRI study when the neck is flexed. MR studies in flexion show not only the anterior displacement of the posterior wall but also a well-enhanced crescent-shaped lesion in the posterior epidural space of the lower cervical canal. This lesion typically disappears when the neck returns to a neutral position, confirming it to be a congested posterior internal vertebral venous plexus rather than a vascular malformation.[
CONCLUSION
We concluded that SSEP did not turn out to be an electrophysiological marker in our HD patients requiring more studies to investigate its significance. Neck flexion did not have an influence on any SSEP parameters in patients or controls.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Abraham A, Gotkine M, Drory VE, Blumend SC. Effect of neck flexion on somatosensory and motor evoked potentials in Hirayama disease. J Neurol Sci. 2013. 334: 102-5
2. Ammendola A, Gallo A, Iannaccone T, Tedeschi G. Hirayama disease: Three cases assessed by F wave, somatosensory and motor evoked potentials and magnetic resonance imaging not supporting flexion myelopathy. Neurol Sci. 2008. 29: 303-11
3. Anuradha S, Fanai V. Hirayama disease: A rare disease with unusual features. Case Rep Neurol Med. 2016. 2016: 5839761
4. Chen T, Hung C, Hsieh T, Lu S, Yang S, Jong Y. Symmetric atrophy of bilateral distal upper extremities and hyperigeaemia in a male adolescent with hirayama disease. J Child Neurol. 2010. 25: 371-4
5. Diluca P, Giraldo H, Muñoz R, Salvatico R, Lambre H, Lylyk P. Magnetic resonance of the cervical spine with sequence in flexion for the diagnosis of Hirayama disease. Rev Argent Radiol. 2017. 81: 105-9
6. Guglielmo GD, Brahe C, Di Muzio A, Uncini A. Benign monomelic amyotrophies of upper and lower limb are not associated to deletions of survival motor neuron gene. J Neurol Sci. 1996. 141: 111-3
7. Hirayama K, Tomonaga M, Kitano K, Yamada T, Kojima S, Arai K. Focal cervical poliopathy causing juvenile muscular atrophy of distal upper extremity: A pathological study. J Neurol Neurosurg Psychiatry. 1987. 50: 285-90
8. Hirayama K. Juvenile muscular atrophy of distal upper extremity (Hirayama disease). Intern Med. 2000. 39: 283-90
9. Hirayama K. Juvenile muscular atrophy of distal upper extremity (Hirayama disease): Focal cervical ischemic poliomyelopathy. Neuropathology. 2000. 20: S91-4
10. Kikuchi S, Tashiro K, Kitagawa M, Iwasaki Y, Abe H. A mechanism of juvenile muscular atrophy localized in the hand and forearm (Hirayama’s disease)-flexion myelopathy with tight dural canal in flexion. Rinsho Shinkeigaku. 1987. 27: 412-9
11. Kim J, Kim Y, Kim S, Oh K. Hirayama disease with proximal involvement. J Korean Med Sci. 2016. 31: 1664-7
12. Kira J, Ochi H. Juvenile muscular atrophy of the distal upper limb (Hirayama disease) associated with atopy. J Neurol Neurosurg Psychiatry. 2001. 70: 798-801
13. Kuwabara S, Nakajima M, Hattori T, Hirayama K. Electrophysiology of juvenile muscular atrophy of unilateral upper limb (Hirayama’s disease). Rinsho Shinkeigaku. 1999. 39: 508-12
14. Lyu RK, Huang YC, Wu YR, Kuo HC, Ro LS, Chen CM. Electrophysiological features of Hirayama disease. Muscle Nerve. 2011. 44: 185-90
15. Martínez HR, Caro-Osorio E, Gutiérrez-Jiménez E, MorenoCuevas J, González-Garza MT. Monomyelic amyotrophy. Rev Mex Neurocienc. 2008. 9: 70-3
16. Mauguiere F, Allison T, Babiloni C, Buchner H, Eisen AA, Goodin DS. Somatosensory evoked potentials. The international federation of clinical neurophysiology. Electroencephalogr Clin Neurophysiol Suppl. 1999. 52: 79-90
17. Misra UK, Kalita J, Mishra VN, Kesari A, Mittal B. A clinical, magnetic resonance imaging, and survival motor neuron gene deletion study of Hirayama disease. Arch Neurol. 2005. 62: 120-3
18. Misra UK, Kalita J, Mishra VN, Phadke RV, Hadique A. Effect of neck flexion on F wave, somatosensory evoked potentials, and magnetic resonance imaging in Hirayama disease. J Neurol Neurosurg Psychiatry. 2006. 77: 695-8
19. Nascimento OJ, Freitas MR. Non-progressive juvenile spinal muscular atrophy of the distal upper limb (Hirayama’s disease): A clinical variant of the benign monomelic amyotrophy. Arq Neuropsiquiatr. 2000. 58: 814-9
20. Park JS, Ko JY, Park D. The reversible effect of neck flexion on the somatosensory evoked potentials in patients with Hirayama disease: A preliminary study. Neurol Sci. 2019. 40: 181-6
21. Restuccia D, Rubino M, Valeriani M, Mirabella M, Sabatelli M, Tonali P. Cervical cord dysfunction during neck flexion in Hirayama’s disease. Neurology. 2003. 60: 1980-3

