

- Department of Neurosurgery, Baylor College of Medicine, Texas Children's Hospital, Houston, TX 77030, USA
Correspondence Address:
Sandi Lam
Department of Neurosurgery, Baylor College of Medicine, Texas Children's Hospital, Houston, TX 77030, USA
DOI:10.4103/2152-7806.178570
Copyright: © 2016 Surgical Neurology International This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Reddy GD, Hansen D, Patel A, Lin Y, Jea A, Lam S. Treatment options for pediatric craniopharyngioma. Surg Neurol Int 11-Mar-2016;7:
How to cite this URL: Reddy GD, Hansen D, Patel A, Lin Y, Jea A, Lam S. Treatment options for pediatric craniopharyngioma. Surg Neurol Int 11-Mar-2016;7:. Available from: http://surgicalneurologyint.com/surgicalint_articles/treatment-options-for-pediatric-craniopharyngioma/
Abstract
Keywords: Adamantinomatous, brain tumor, craniopharyngioma, pediatric
ILLUSTRATIVE CASES
Case 1
A 13-year-old female with intermittent headaches evaluated by an ophthalmologist was noted to have a retinal abnormality, prompting a magnetic resonance imaging (MRI) scan and referral to the neurosurgery service. On initial exam, the patient was neurologically intact and without headache. Imaging revealed a complex heterogeneous cystic mass, arising from a suprasellar location, invading into the third ventricle, and closely apposed to the hypothalamus bilaterally. There was mild contrast enhancement peripherally and inferiorly. Of note, the initial MRI and clinical presentation [
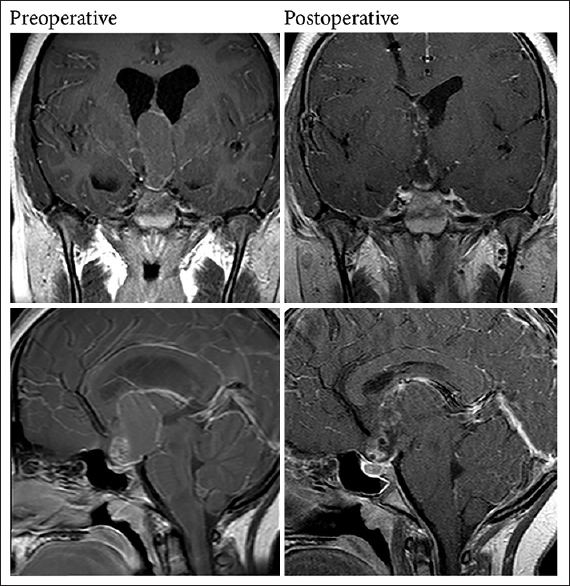
Case 2
A 13-year-old female presented with headache, fatigue, and nausea. On exam, she was neurologically intact though with severe headache. Imaging revealed a large, complex, heterogeneous cystic mass, arising from a suprasellar location, invading into the third ventricle, and closely apposed to the hypothalamus bilaterally. She had significant hydrocephalus resulting from obstruction of the third ventricle. An external ventricular drain was placed, followed by endoscopic fenestration of the cyst, biopsy of the mass, and placement of a ventriculoperitoneal shunt. Pathology from the biopsy confirmed the suspected diagnosis of craniopharyngioma. The following year, an Ommaya reservoir was placed into an enlarging suprasellar cyst for intermittent as-needed drainage of accumulating fluid. Postoperative imaging showed a decompressed suprasellar cyst; she continues with expectant management of an inferior prepontine cyst [
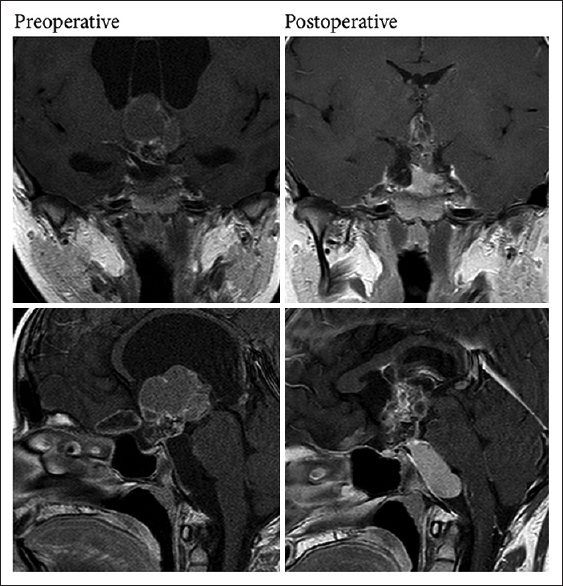
EPIDEMIOLOGY AND CLINICAL PRESENTATION
Craniopharyngiomas are rare tumors, with an estimated incidence of 2–3/1 million. They are most common in children (age 5–15 years old) and older adults (60–70 years of age).[
These tumors are typically located in or above the sella turcica and produce symptoms by compression of adjacent neural structures. Slow growth and insidious onset of symptoms often delay arriving at a diagnosis. Potential symptoms are wide-ranging. They include visual deficits from compression of the optic apparatus, endocrine deficiencies, such as diabetes insipidus (DI) or pan-hypopituitarism from compression of the pituitary gland or stalk, hypothalamic compression and dysfunction resulting in abnormalities in sleep, appetite, or thermal regulation, or symptoms of hydrocephalus such as headache or vomiting from obstruction of cerebral spinal fluid pathways.[
TUMOR BIOLOGY
Craniopharyngiomas occur in two histological subtypes: An adamantinomatous form that is the most common pediatric variant and a papillary form that is found almost exclusively in adults. The pediatric form is thought to arise from epithelial remnants of the craniophayngeal duct or Rathke's pouch, an embryologic structure that develops into the anterior pituitary. These remnants are thought to enlarge during the development of the pituitary gland and thus present early in life. Grossly, these tumors typically have both solid and cystic components and are often calcified on imaging. The cyst fluid is dark, oily, and rich in lipids with birefringent cholesterol crystals.[
Recent genetic analysis has also shown differences between these two subtypes. Mutations in B-catenin (CTNNB1), a downstream effector of the Wnt pathway that is, involved in cellular growth and development, has been described in 60–96% of adamantinomatous craniopharyngiomas.[
MEDICAL WORKUP AND MANAGEMENT
A complete workup for craniopharyngioma should include MRI with and without gadolinium contrast to characterize the tumor and its relationship with critical nearby neural structures. As demonstrated in Case 1, these tumors may microscopically invade these structures even when this is not apparent on preoperative imaging. MR angiography can also be helpful in delineating the location of nearby vessels at the skull base. A noncontrasted computed tomography scan can also reveal complex calcifications and expansion of the sella, which is helpful in narrowing the differential. In addition, a workup for endocrinopathies should also be performed with measurements of growth hormone, thyroid stimulating hormone, follicular stimulating hormone/luteinizing hormone, prolactin, cortisol, and serum electrolytes. Any abnormalities ideally should be corrected before surgery is performed. Finally, formal visual acuity and visual field assessment are important to characterize any deficits that exist preoperatively.
TREATMENT AND OUTCOMES
Although histologically benign, these tumors frequently recur after treatment, and their close association with critical neurologic structures can lead to a much more malignant course. Surgical treatment options range from GTR to more conservative surgery (i.e., subtotal resection [STR] or biopsy only) followed by postoperative radiotherapy (RT), or other less invasive procedures such as endoscopic cyst fenestration or placement of an Ommaya reservoir into the tumor cyst for delivery of antineoplastic agents.[
Historically, open cranial surgery with the goal of achieving GTR has been the treatment of choice, as it allows rapid decompression, provides a histological diagnosis, and is thought to minimize recurrence. The results of GTR have been influenced largely by surgeon experience, and the tendency of these tumors to invade nearby critical neuromuscular structures often leads to significant morbidity.[
The lack of acceptable outcomes with GTR has led to groups approaching these tumors with a more conservative surgical plan, including STR or biopsy, followed in some cases with RT.[
For the surgeon experienced in endoscopy, an endoscopic endonasal approach, as compared to an open approach, has been shown to provide higher rates for GTR (66–69% vs. 48%), with lower reoccurrence (18% vs. 28%), with lower rates of permanent DI (27–32% vs. 48%), and less visual deterioration (1.7% vs. 11%).[
Perhaps the least invasive treatment option is the insertion of an Ommaya reservoir into the cystic aspect of the tumor followed by drainage with or without subsequent instillation of antineoplastic agents. Several reports have shown follow-up out to 7 years with good cyst size control and 43–73% of patients needing no additional treatment.[
Radiation therapy, either as a first-line treatment or as an adjuvant to surgical resection, has become a frequent care option. SRS can deliver radiation with a steeper dose gradient between tumor and adjacent brain structures. It is believed to lead to lower rates of neurotoxic side effects in comparison to traditional fractionated RT. The potential side effects of radiation are similar to that of open surgery and include the following: Panhypopituitarism, DI, hypothalamic dysfunction, vasculopathy, cognitive dysfunction, optic neuropathy, and secondary malignancies.[
Finally, systemic chemotherapy treatment with INF-alpha-2b has been performed by the Pediatric Brain Tumor Consortium (PBTC-039) phase 2 trial in pediatric patients with recurrent craniopharyngioma. Three of the 12 patients tested experienced a response to the drug, and none developed any permanent side effects.[
CHOICE OF THERAPY
The treatment paradigm for craniopharyngioma has evolved over time, as our treatment options have expanded and our understanding of the long-term consequences of radical resection has grown. Originally, aggressive resection was the only hope of controlling this tumor; in spite of the often severe morbidity of such an approach, it remained the mainstay of treatment. But as the natural history of this tumor is recurrence, it may be considered a chronic disease, with the goal of maximizing control but minimizing patient morbidity. Our desire as surgeons to obtain tumor-free postoperative imaging may overlook the impact on our patient. Weighing risks, benefits, and alternatives to surgical goals and approaches are crucial in the treatment of this challenging tumor.
To date, no class I recommendations exist for the best treatment of these tumors. Management should be by a multidisciplinary team (neurosurgery, endocrinology, ophthalmology, psychology, oncology, and radiation oncology) and be individualized for each patient. There are some practical surgical considerations to keep in mind. In cases where total resection can be obtained without significant morbidity (i.e., cases where the tumor is not invading or adherent to the hypothalamus), GTR remains the treatment of choice. In cases where the tumor is small and the solid portions are primary intrasellar, without significant extension laterally in the suprasellar space or without encasement of vessels, an endoscopic approach may provide good outcomes. In cases where the tumor is densely involved in critical structures and has a significant cystic component causing mass effect, an Ommaya placement with or without chemotherapy infusion may represent a less invasive way to decompress neural structures and control tumor progression. Finally, in cases of STR or recurrent disease, particularly with a favorable margin between the tumor and the optic chiasm, adjuvant RT or SRS is likely to improve progression-free survival. Promising treatments from PBTC trials may offer hope for future therapies with lower side effect profiles. Ultimately, this tumor remains one of the most difficult pediatric neurosurgical problems, and recommendations will continue to evolve.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
1. Adler JR, Gibbs IC, Puataweepong P, Chang SD. Visual field preservation after multisession cyberknife radiosurgery for perioptic lesions. Neurosurgery. 2006. 59: 244-54
2. Agha A, Sherlock M, Brennan S, O’Connor SA, O’sullivan E, Rogers B. Hypothalamic-pituitary dysfunction after irradiation of nonpituitary brain tumors in adults. J Clin Endocrinol Metab. 2005. 90: 6355-60
3. Ali ZS, Bailey RL, Daniels LB, Vakhshori V, Lewis DJ, Hossain AT. Comparative effectiveness of treatment options for pediatric craniopharyngiomas. J Neurosurg Pediatr. 2014. 13: 178-88
4. Brastianos PK, Taylor-Weiner A, Manley PE, Jones RT, Dias-Santagata D, Thorner AR. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat Genet. 2014. 46: 161-5
5. Buslei R, Nolde M, Hofmann B, Meissner S, Eyupoglu IY, Siebzehnrübl F. Common mutations of beta-catenin in adamantinomatous craniopharyngiomas but not in other tumours originating from the sellar region. Acta Neuropathol. 2005. 109: 589-97
6. Caldarelli M, Massimi L, Tamburrini G, Cappa M, Di Rocco C. Long-term results of the surgical treatment of craniopharyngioma: The experience at the Policlinico Gemelli, Catholic University, Rome. Childs Nerv Syst. 2005. 21: 747-57
7. Cavalheiro S, Di Rocco C, Valenzuela S, Dastoli PA, Tamburrini G, Massimi L. Craniopharyngiomas: Intratumoral chemotherapy with interferon-alpha: A multicenter preliminary study with 60 cases. Neurosurg Focus. 2010. 28: E12-
8. Clark AJ, Cage TA, Aranda D, Parsa AT, Sun PP, Auguste KI. A systematic review of the results of surgery and radiotherapy on tumor control for pediatric craniopharyngioma. Childs Nerv Syst. 2013. 29: 231-8
9. Cohen M, Bartels U, Branson H, Kulkarni AV, Hamilton J. Trends in treatment and outcomes of pediatric craniopharyngioma, 1975-2011. Neuro Oncol. 2013. 15: 767-74
10. Darzy KH, Shalet SM. Hypopituitarism after cranial irradiation. J Endocrinol Invest. 2005. 28: 78-87
11. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro Oncol. 2012. 14: v1-49
12. Duff J, Meyer FB, Ilstrup DM, Laws ER, Schleck CD, Scheithauer BW. Long-term outcomes for surgically resected craniopharyngiomas. Neurosurgery. 2000. 46: 291-302
13. Elliott RE, Hsieh K, Hochm T, Belitskaya-Levy I, Wisoff J, Wisoff JH. Efficacy and safety of radical resection of primary and recurrent craniopharyngiomas in 86 children. J Neurosurg Pediatr. 2010. 5: 30-48
14. Fahlbusch R, Honegger J, Paulus W, Huk W, Buchfelder M. Surgical treatment of craniopharyngiomas: Experience with 168 patients. J Neurosurg. 1999. 90: 237-50
15. Garnett MR, Puget S, Grill J, Sainte-Rose C. Craniopharyngioma. Orphanet J Rare Dis. 2007. 2: 18-
16. Garrè ML, Cama A. Craniopharyngioma: Modern concepts in pathogenesis and treatment. Curr Opin Pediatr. 2007. 19: 471-9
17. Gautier A, Godbout A, Grosheny C, Tejedor I, Coudert M, Courtillot C. Markers of recurrence and long-term morbidity in craniopharyngioma: A systematic analysis of 171 patients. J Clin Endocrinol Metab. 2012. 97: 1258-67
18. Habrand JL, Ganry O, Couanet D, Rouxel V, Levy-Piedbois C, Pierre-Kahn A. The role of radiation therapy in the management of craniopharyngioma: A 25-year experience and review of the literature. Int J Radiat Oncol Biol Phys. 1999. 44: 255-63
19. Jakacki RI, Cohen BH, Jamison C, Mathews VP, Arenson E, Longee DC. Phase II evaluation of interferon-alpha-2a for progressive or recurrent craniopharyngiomas. J Neurosurg. 2000. 92: 255-60
20. Julow J, Backlund EO, Lányi F, Hajda M, Bálint K, Nyáry I. Long-term results and late complications after intracavitary yttrium-90 colloid irradiation of recurrent cystic craniopharyngiomas. Neurosurgery. 2007. 61: 288-95
21. Karavitaki N, Brufani C, Warner JT, Adams CB, Richards P, Ansorge O. Craniopharyngiomas in children and adults: Systematic analysis of 121 cases with long-term follow-up. Clin Endocrinol (Oxf). 2005. 62: 397-409
22. Kato K, Nakatani Y, Kanno H, Inayama Y, Ijiri R, Nagahara N. Possible linkage between specific histological structures and aberrant reactivation of the Wnt pathway in adamantinomatous craniopharyngioma. J Pathol. 2004. 203: 814-21
23. Kobayashi T, Kida Y, Mori Y, Hasegawa T. Long-term results of gamma knife surgery for the treatment of craniopharyngioma in 98 consecutive cases. J Neurosurg. 2005. 103: 482-8
24. Komotar RJ, Starke RM, Raper DM, Anand VK, Schwartz TH. Endoscopic endonasal compared with microscopic transsphenoidal and open transcranial resection of craniopharyngiomas. World Neurosurg. 2012. 77: 329-41
25. Manley PE, McKendrick K, McGillicudy M, Chi SN, Kieran MW, Cohen LE. Sleep dysfunction in long term survivors of craniopharyngioma. J Neurooncol. 2012. 108: 543-9
26. McCord MW, Buatti JM, Fennell EM, Mendenhall WM, Marcus RB, Rhoton AL. Radiotherapy for pituitary adenoma: Long-term outcome and sequelae. Int J Radiat Oncol Biol Phys. 1997. 39: 437-44
27. Minniti G, Esposito V, Amichetti M, Enrici RM. The role of fractionated radiotherapy and radiosurgery in the management of patients with craniopharyngioma. Neurosurg Rev. 2009. 32: 125-32
28. Moussa AH, Kerasha AA, Mahmoud ME. Surprising outcome of ommaya reservoir in treating cystic craniopharyngioma: A retrospective study. Br J Neurosurg. 2013. 27: 370-3
29. Müller HL. Childhood craniopharyngioma. Pituitary. 2013. 16: 56-67
30. Müller HL. Craniopharyngioma - A childhood and adult disease with challenging characteristics. Front Endocrinol (Lausanne). 2012. 3: 80-
31. Samii M, Tatagiba M. Surgical management of craniopharyngiomas: A review. Neurol Med Chir (Tokyo). 1997. 37: 141-9
32. Schoenfeld A, Pekmezci M, Barnes MJ, Tihan T, Gupta N, Lamborn KR. The superiority of conservative resection and adjuvant radiation for craniopharyngiomas. J Neurooncol. 2012. 108: 133-9
33. Sekine S, Shibata T, Kokubu A, Morishita Y, Noguchi M, Nakanishi Y. Craniopharyngiomas of adamantinomatous type harbor beta-catenin gene mutations. Am J Pathol. 2002. 161: 1997-2001
34. Steinbok P, Hukin J. Intracystic treatments for craniopharyngioma. Neurosurg Focus. 2010. 28: E13-
35. Weiner HL, Wisoff JH, Rosenberg ME, Kupersmith MJ, Cohen H, Zagzag D. Craniopharyngiomas: A clinicopathological analysis of factors predictive of recurrence and functional outcome. Neurosurgery. 1994. 35: 1001-10
36. Wisoff J, Donahue B, Albright AL, Pollack IF, Adelson PD.editors. Craniopharyngiomas. Principles and Practice of Pediatric Neurosurgery. New York: Thieme; 2007. p. 560-78
37. Yasargil MG, Curcic M, Kis M, Siegenthaler G, Teddy PJ, Roth P. Total removal of craniopharyngiomas. Approaches and long-term results in 144 patients. J Neurosurg. 1990. 73: 3-11
38. Yeung JT, Pollack IF, Panigrahy A, Jakacki RI. Pegylated interferon-a-2b for children with recurrent craniopharyngioma. J Neurosurg Pediatr. 2012. 10: 498-503

