

- Department of Neurological Surgery, Fundación Universitaria de Ciencias de la Salud, Hospital de San José - Sociedad de Cirugía de Bogotá, Bogotá, Colombia,
- School of Medicine, Universidad Nacional de Colombia, Bogotá, Colombia,
- Research Institute, Fundación Universitaria de Ciencias de la Salud, Bogotá, Colombia,
- Department of Neurosurgery, San Fernando Hospital, San Fernando, Argentina,
- Departament of Microbiology, Universidad Nacional de Colombia, Bogotá, Colombia
- Translational Research Group in Oncology, Instituto Nacional de Cancerología, Bogotá, Colombia
- Academic direction, Universidad Nacional de Colombia - Sede de La Paz, La Paz, Colombia
- Department of Neurosurgery, Hospital Universitario Fundación Santa Fé de Bogotá, Bogotá, Colombia,
- Department of Psychiatry, New York University Langone Health, New York City, USA,
- 0Department of Anatomy, University of Buenos Aires, Buenos Aires, Argentina,
- 1Department of Neurosurgery, Instituto de Seguridad y Servicios Sociales de los Trabajadores del Estado, Mexico City, Mexico,
- 2Department of Neurosurgery, University of Pavia, Polo Didattico “Cesare Brusotti”, Pavia, Italy,
- 3Hospital Padilla de Tucuman, Tucuman, Argentina.
- 4Department of Pathology, Instituto Nacional de Cancerología Bogotá, Bogotá, Colombia.
Correspondence Address:
Edgar G. Ordóñez-Rubiano, Department of Neurological Surgery, Fundación Universitaria de Ciencias de la Salud, Hospital de San José - Sociedad de Cirugía de Bogotá, Colombia.
DOI:10.25259/SNI_209_2023
Copyright: © 2023 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Edgar G. Ordóñez Rubiano1,2,3, Matías Baldoncini4, Alba Lucía Cómbita5,6, César Payán-Gómez7, Diego F. Gómez-Amarillo8, Fernando Hakim8, Luisa Fernanda Figueredo9, Valeria Forlizzi10, Carlos Castillo Rangel11, Sabino Luzzi12, Alvaro Campero13, Rafael Parra-Medina3,14. Understanding the molecular profiling of diffuse gliomas classification: A brief overview. 30-Jun-2023;14:225
How to cite this URL: Edgar G. Ordóñez Rubiano1,2,3, Matías Baldoncini4, Alba Lucía Cómbita5,6, César Payán-Gómez7, Diego F. Gómez-Amarillo8, Fernando Hakim8, Luisa Fernanda Figueredo9, Valeria Forlizzi10, Carlos Castillo Rangel11, Sabino Luzzi12, Alvaro Campero13, Rafael Parra-Medina3,14. Understanding the molecular profiling of diffuse gliomas classification: A brief overview. 30-Jun-2023;14:225. Available from: https://surgicalneurologyint.com/surgicalint-articles/12386/
Date of Submission
04-Mar-2023
Date of Acceptance
04-Jun-2023
Date of Web Publication
30-Jun-2023
Abstract
Background: Gliomas represent almost 30% of all primary brain tumors and account for 80% of malignant primary ones. In the last two decades, significant progress has been made in understanding gliomas’ molecular origin and development. These advancements have demonstrated a remarkable improvement in classification systems based on mutational markers, which contribute paramount information in addition to traditional histology-based classification.
Methods: We performed a narrative review of the literature including each molecular marker described for adult diffuse gliomas used in the World Health Organization (WHO) central nervous system 5.
Results: The 2021 WHO classification of diffuse gliomas encompasses many molecular aspects considered in the latest proposed hallmarks of cancer. The outcome of patients with diffuse gliomas relies on their molecular behavior and consequently, to determine clinical outcomes for these patients, molecular profiling should be mandatory. At least, the following molecular markers are necessary for the current most accurate classification of these tumors: (1) isocitrate dehydrogenase (IDH) IDH-1 mutation, (2) 1p/19q codeletion, (3) cyclin-dependent kinase inhibitor 2A/B deletion, (4) telomerase reverse transcriptase promoter mutation, (5) α-thalassemia/ mental retardation syndrome X-linked loss, (6) epidermal growth factor receptor amplification, and (7) tumor protein P53 mutation. These molecular markers have allowed the differentiation of multiple variations of the same disease, including the differentiation of distinct molecular Grade 4 gliomas. This could imply different clinical outcomes and possibly impact targeted therapies in the years to come.
Conclusion: Physicians face different challenging scenarios according to the clinical features of patients with gliomas. In addition to the current advances in clinical decision-making, including radiological and surgical techniques, understanding the disease’s molecular pathogenesis is paramount to improving the benefits of its clinical treatments. This review aims to describe straightforwardly the most remarkable aspects of the molecular pathogenesis of diffuse gliomas.
Keywords: Classification, Diffuse glioma, Glioblastoma, Glioma, Molecular pathology
INTRODUCTION
Gliomas represent about 30% of all primary brain tumors and are responsible for the majority of primary brain tumor deaths.[
In different scenarios, health-care providers of different specialties are involved in the treatment of patients with gliomas and should be familiarized with terminology regarding patients’ tumor diagnoses. Neurooncologists and neurologists as well as neurosurgeons should understand not only the terminology of molecular profiling but also the biological meaning and the implications behind these terms. In this review, we aim to describe and summarize the most remarkable aspects of the molecular pathogenesis of adult diffuse gliomas.
MATERIALS AND METHODS
The authors performed a narrative review of the literature including each molecular marker described for adult diffuse gliomas used in the WHO CNS5. A MEDLINE/PUBMED search was based on the references of retrieved literature. This review included a comprehensive overview of the 3rd, 4th, and 5th editions of the WHO classification.
RESULTS
Histopathological classification
According to the WHO CNS3, gliomas include astrocytomas of various grades (diffuse astrocytoma [Grades II and III] and GBM [Grade IV]), oligodendrogliomas (Grades II and III), and mixed oligoastrocytomas (Grades II and III) [
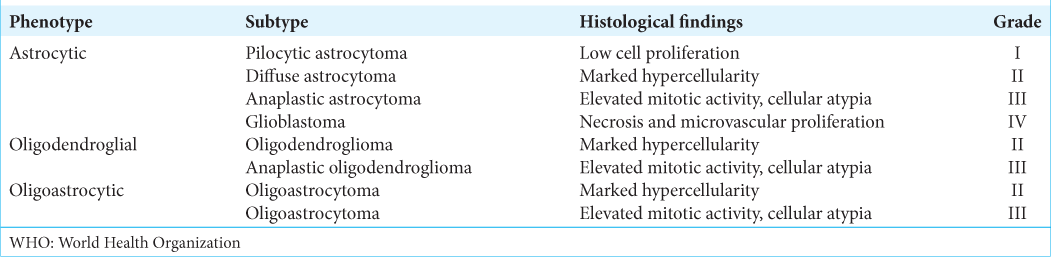
Molecular classification
The WHO Grade II–III gliomas are the most prevalent gliomas in young adults and are characterized by diffuse infiltration of the brain and their proclivity for recurring and malignant progression. Histological classification had previously distinguished astrocytic, oligodendroglial, and mixed oligoastrocytic tumors. However, molecular markers have now revealed evidence of only two subtypes: (1) the astrocytic genotype, which is characterized by mutations in tumor protein P53 (TP53), often accompanied by a mutation in the α-thalassemia/mental retardation syndrome X-linked (ATRX) gene, and (2) the oligodendroglial genotype, which is characterized by the codeletion of 1p and 19q chromosomal arms (1p/19q codeletion), associated with the mutation of the telomerase reverse transcriptase (TERT) promoter (TERTp).[
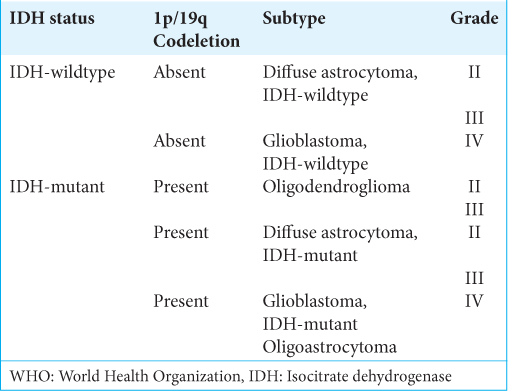
To improve the classification of diffuse gliomas, the WHO CNS5[
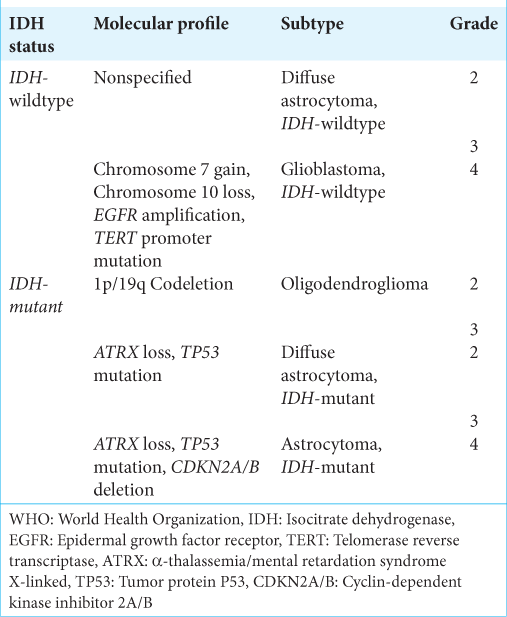
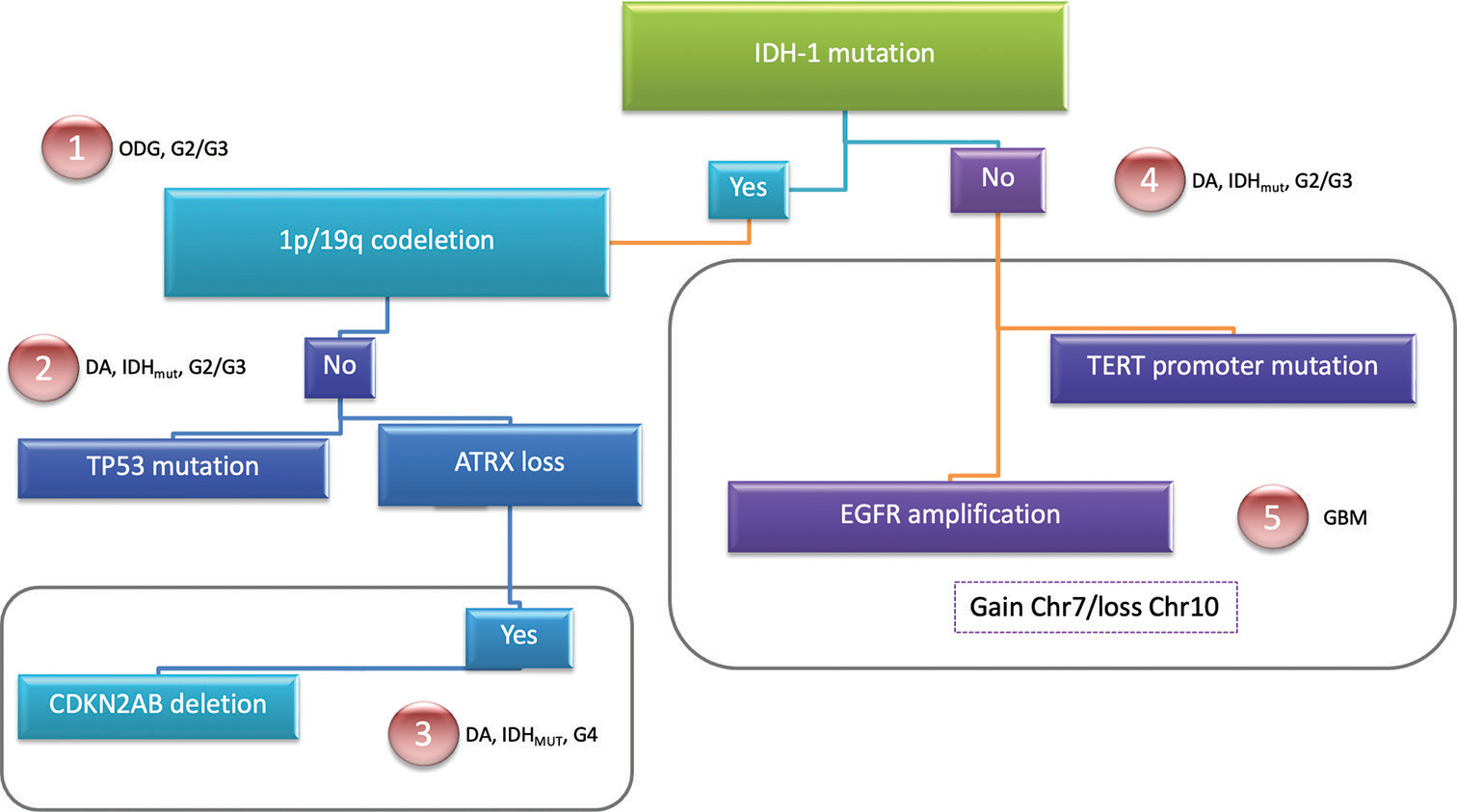
Figure 1:
The World Health Organization Central Nervous System 5. Molecular biomarkers used for classifying diffuse gliomas. A simple resume for easy differentiation of the presence of mutations in each entity is demonstrated. Sequential acquisition of markers can be performed to perform a correct diagnosis. The five different subtypes of diffuse gliomas are shown. DA: Diffuse astrocytoma, ODG: Oligodendroglioma, GBM: Glioblastoma, IDHmut: Isocitrate dehydrogenase mutated, ATRX: Alfa-thalassemia/mental retardation syndrome X-linked, CDKN2AB: Cyclin-dependent kinase inhibitor 2A/B, EGFR: Epidermal growth factor receptor, TERT: Telomerase reverse transcriptase.
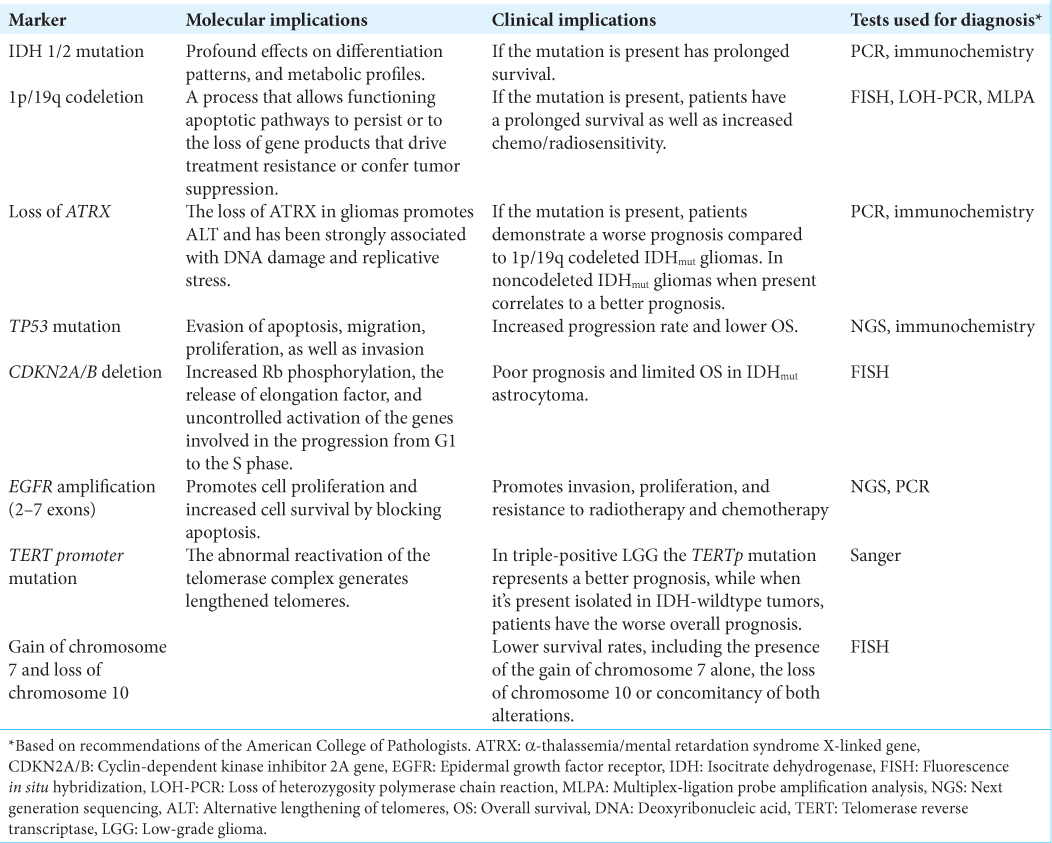
Hallmarks of cancer: Diffuse gliomas
As the global tendency is to characterize and classify tumors by their molecular behavior, it is of major importance to enhance the intrinsic properties of tumors described in the last decade. All gliomas, but more specifically GBM are recognized by their malignant behavior and their complex molecular biology. To understand these malignant properties, it is fundamental to comprehend the basis of cancer. Hanahan and Weinberg proposed the “Hallmarks of Cancer” as a set of functional capabilities that make cells their way from normalcy to neoplastic growth states, which are characteristics that are basic for their ability to form cancer.[
Core mutational markers
Core mutational markers have been described for diffuse gliomas given the importance of differentiating separate entities in terms of biological behavior and the influence on clinical prognosis. These markers have been identified in multiple studies demonstrating the presence or not in each different glioma subtype. Hereafter, we resume the most valuable information regarding these core molecular markers.
IDH-1 mutation
Diffuse gliomas of both genotypes can carry mutations in the gene for the enzyme IDH 1 (IDH1) [
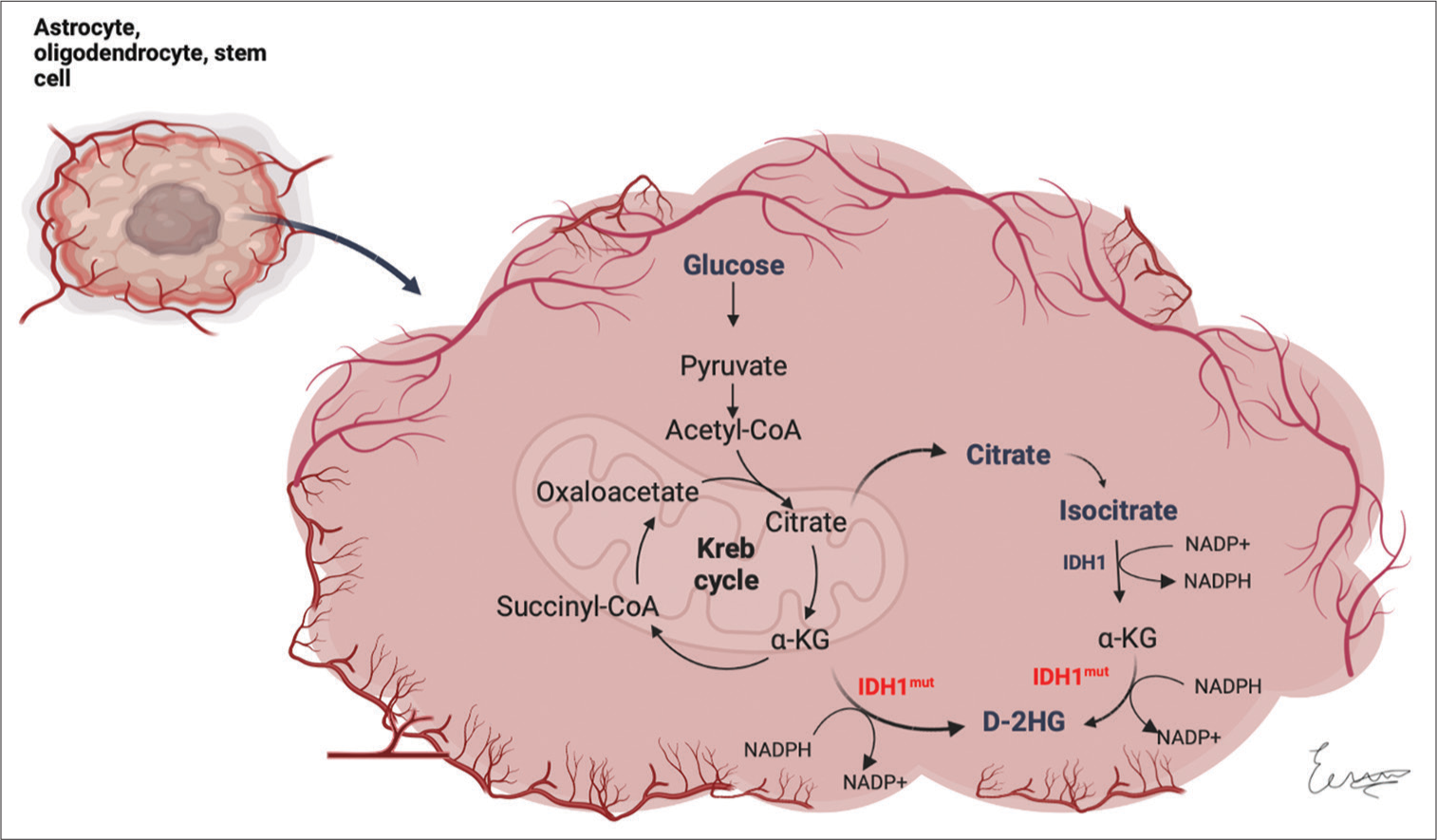
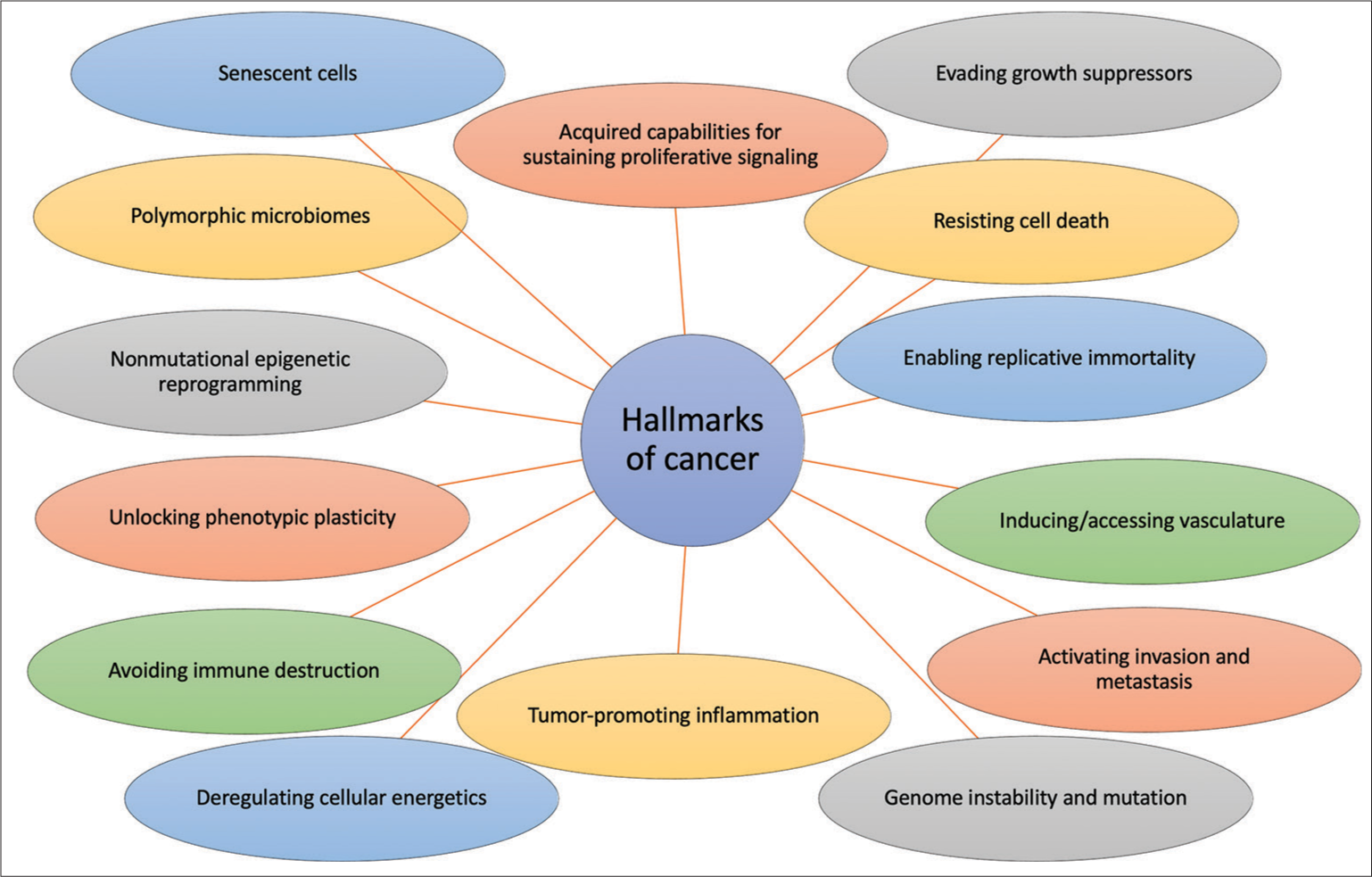
Figure 3:
Impairment of cellular metabolism in IDH mutant diffuse gliomas. The enzyme isocitrate dehydrogenase 1 normally catalyzes the oxidative decarboxylation of isocitrate to form 2-oxoglutarate (α-KG). When the enzyme is mutated, it shows a gain in function to produce D-2-hydroxyglutarate from α-KG. α-KG: Alpha-ketoglutarate, IDH1: Isocitrate dehydrogenase 1, IDH1mut: Mutated isocitrate dehydrogenase 1, D-2HG: D-2 hydroxyglutarate. (Created with
IDH enzymes normally catalyze the oxidative decarboxylation of isocitrate to 2-oxoglutarate (alpha-ketoglutarate [α-KG]), thus generating nicotinamide adenine dinucleotide phosphate (NADPH) from NADP+. When the gene is mutated, the enzyme shows a gain in function to produce D-2-hydroxyglutarate (D-2HG) from α-KG, demonstrating a decreased NADPH production, which probably predisposes cells to oxidative stress.[
1p/19q codeletion
Reifenberger et al. were the first in demonstrating that oligodendroglial tumors show allelic deletions on 19q (deletions in different regions of the short arm of chromosome 19 with similar phenotype) and the majority of tumors with 19q loss also show loss of alleles on lp.[
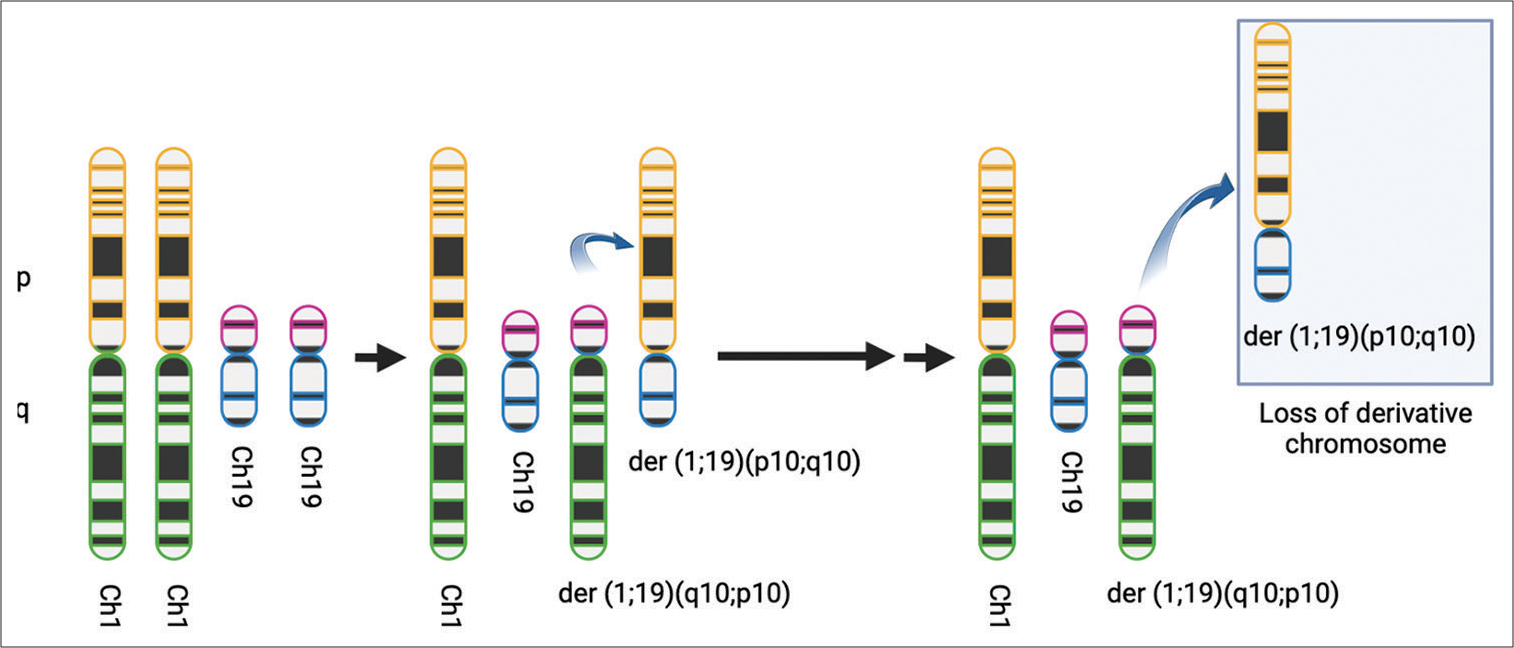
Figure 4:
Codeletion of 1p/19q. It is believed that an unbalanced whole-arm translocation between chromosomes 1 and 19 creates two derivatives chromosomes: der (1;19) (q10;p10) and der (1;19) (p10;q10). This translocation generates the loss of derivative chromosome der (1;19) (p10;q10), which contains 1p and 19q. (Created with
Loss of ATRX
The role of the ATRX gene in cancer has emerged in the past decade. The status of the ATRX is now pivotal for glioma classification and has been included as part of the molecular profile for glioma characterization.[
TP53 mutation
TP53 is a tumor suppressor. Normal TP53 protein functions as an inhibitor of cellular replication when sustained DNA damage and its gene mutation leads to an impaired protein that alters its ability to bind DNA and prevent cell replication.[
CDKN2A/B homozygous deletion
According to the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy — Not Official WHO (cIMPACT-NOW) recommendation,[
CDKs are core regulatory molecules that determine cellular progression through the cell cycle. Extracellular signals lead to complex formation between D-type cyclins and cyclin-dependent kinases 4 and 6 (CDK4/CDK6) [
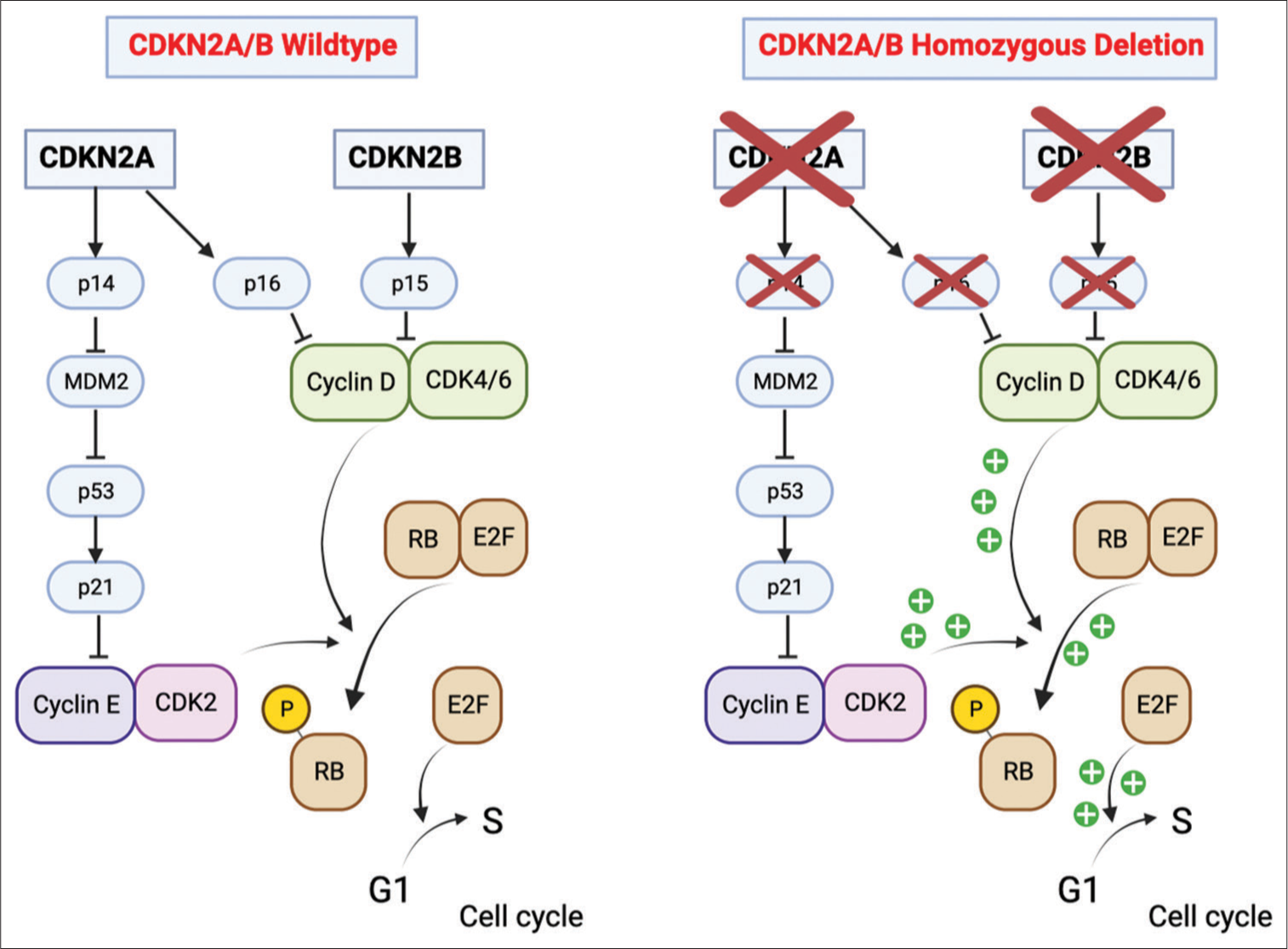
Figure 5:
Cyclin-dependent kinase inhibitor 2A/B (CDKN2A/B) homozygous deletion. In nonmutated cells, the CDKN2A gene synthesizes p16 and p14. p16 binds to and inactivates CDK4/6. The CDKN2B gene is adjacent to CDKN2A and encodes p15, which inactivates CDK4/6. The p14 protein is an alternate reading frame protein and prevents p53 degradation by inhibiting MDM2 activity. This generates the activation of p21, which binds to and inhibits the activity of CDK2. By binding to these cyclin-dependent kinases, p14, p15, and p16 block cell cycle progression from G1 to the S phase. On the other hand, when CDKN2A/B is homozygously deleted, the cells are unable to synthesize these tumor suppressor proteins. Consequently, there is an unrestricted generation of complexes between cyclins and cyclin-dependent kinases, which leads to increased Rb phosphorylation, a release of elongation factor, and uncontrolled activation of the genes involved in the progression from G1 to the S phase. (Created with
EGFR amplification
EGFR is a cell transmembrane tyrosine kinase receptor, and its gene (EGFR) is in the chromosome band 7p12.1.[
TERT promoter mutation
Telomerase is a protein mainly conformed by human telomerase ribonucleic acid and TERT. The telomerase is located within the cell nucleus, and its function is to synthesize a repetitive nucleotide sequence (TTAGGG) while creating the telomeres at the end of chromosomes to protect genomic DNA.[
Killela et al. reported that TERTp mutations were present in about 80% of GBM and oligodendrogliomas.[
Gain of chromosome 7 and loss of chromosome 10
Gain of chromosome 7 alone has shown an increased recurrence of gliomas in the pediatric population.[
DISCUSSION
Glioma precision medicine
Just before the publication of the WHO CNS5, Weller et al. published the EANO guidelines of adulthood diffuse gliomas.[
In addition to separate all entities, the WHO CNS 5 allows to evaluate prognosis in terms of OS and chemo and radiosensitivity.[
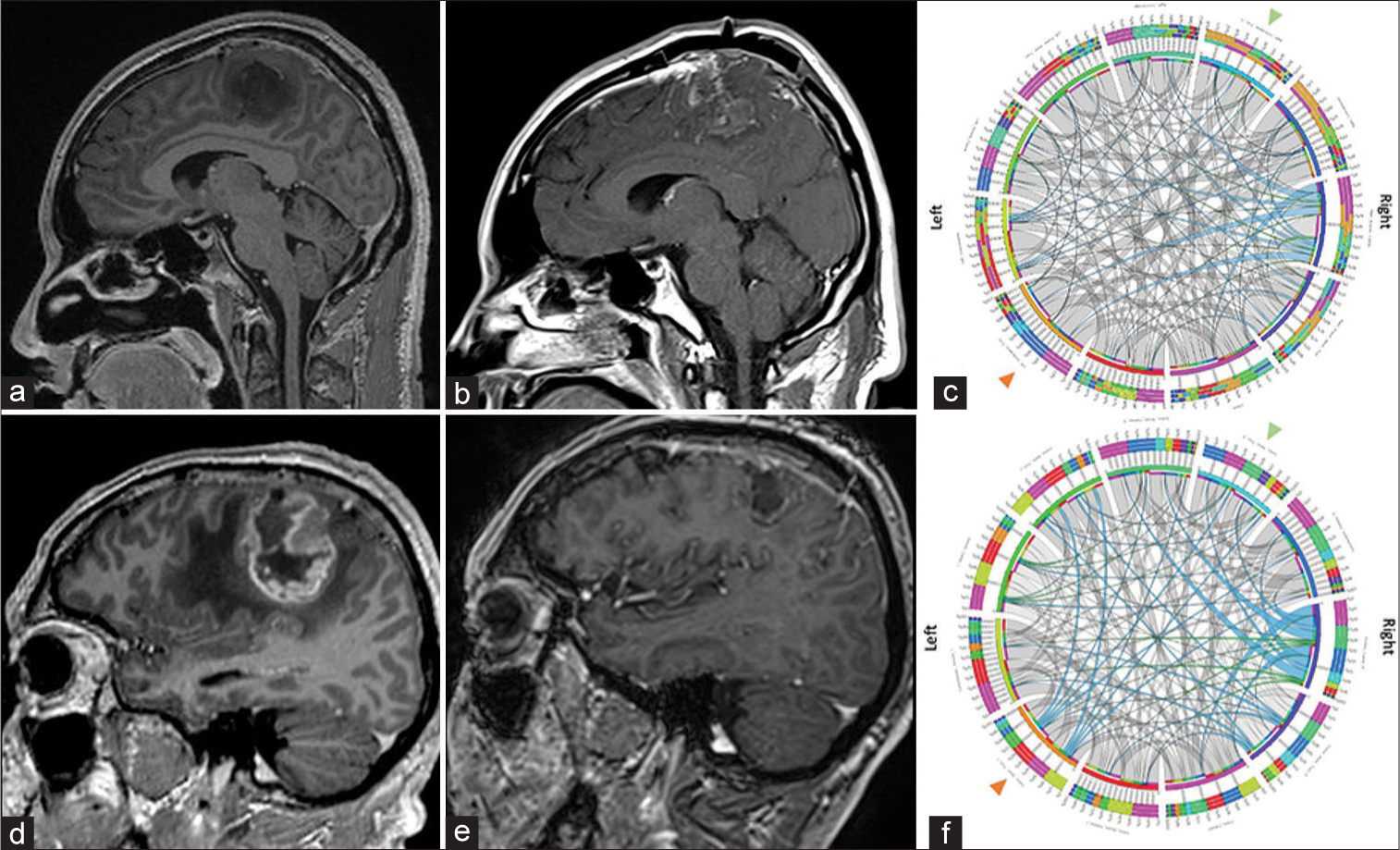
Figure 6:
Neuroimaging of an oligodendroglioma Grade 2 and of an isocitrate dehydrogenase (IDH) IDHmut astrocytoma, Grade 4. Case 1. Oligodendroglioma Grade 2. (a) Preoperative axial T1 postcontrast imaging demonstrating a nonenhancing perirolandic hypointense tumor. (b) Immediate postoperative enhanced magnetic resonance imaging (MRI) demonstrating near total resection of the tumor. (c) Preoperative connectivity map. White fibers are arranged in a symmetric distribution in both, left and right cortical tracts, noting in blue an increased signal diffusion in the right Aslant tract. Green arrowhead: Right corticospinal tract, Orange Arrowhead: Left corticospinal tract. Case 2. IDHmut astrocytoma, grade 4. (d) Preoperative enhanced MRI demonstrates a large enhancing perirolandic tumor consistent with a high-grade glioma. (e) 3-month follow-up MRI demonstrates adequate control with no enhancing tumor. (f) Preoperative connectivity map. White fibers are arranged in a symmetric distribution in both, the left and right cortical tracts, note in blue an increased signal diffusion in the right Aslant tract, the left corticospinal tract, and the right external capsule. Green arrowhead: Right corticospinal tract, Orange Arrowhead: Left corticospinal tract.
CONCLUSION
Here, we resume some of the most important current markers used for the molecular classification of diffuse gliomas. The understanding of the genetic origin of the molecular features of gliomas is mandatory for neurosurgeons and neuro-oncologists. The literature regarding this topic is overwhelming and increases day by day. The complex molecular structure among the various type of gliomas generates the investigation of targets for treatment one of the most challenging tasks for neuroscientists in this era. The most remarkable molecular markers must be included in the study for the diagnosis of these tumors whenever possible to guide in a case-by-case manner the adjuvant treatment. Certainly, differentiating all genetic/molecular subtypes of diffuse gliomas will demonstrate why the prognosis changes are sometimes notable between one case and another, even for tumors in the same molecular group with the same extent of resection rates. Finally, complementary genomic and transcriptomic studies must continue to be performed, as they will help to elucidate the covered origins of molecular cascades and will guide us to a better understanding of these neoplasms.
Declaration of patient consent
Patients’ consent not required as patients’ identities were not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
1. Albertson DG. Gene amplification in cancer. Trends Genet. 2006. 22: 447-55
2. Appay R, Dehais C, Maurage CA, Alentorn A, Carpentier C, Colin C. CDKN2A homozygous deletion is a strong adverse prognosis factor in diffuse malignant IDH-mutant gliomas. Neuro Oncol. 2019. 21: 1519-28
3. Arita H, Narita Y, Fukushima S, Tateishi K, Matsushita Y, Yoshida A. Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta Neuropathol. 2013. 126: 267-76
4. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008. 116: 597-602
5. Bell RJ, Rube HT, Kreig A, Mancini A, Fouse SD, Nagarajan RP. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science. 2015. 348: 1036-9
6. Belotti Y, Tolomeo S, Yu R, Lim WT, Lim CT. Prognostic neurotransmitter receptors genes are associated with immune response, inflammation and cancer hallmarks in brain tumors. Cancers (Basel). 2022. 14: 2544
7. Bieńkowski M, Piaskowski S, Stoczyńska-Fidelus E, Szybka M, Banaszczyk M, Witusik-Perkowska M. Screening for EGFR amplifications with a novel method and their significance for the outcome of glioblastoma patients. PLoS One. 2013. 8: e65444
8. Biernat W, Huang H, Yokoo H, Kleihues P, Ohgaki H. Predominant expression of mutant EGFR (EGFRvIII) is rare in primary glioblastomas. Brain Pathol. 2004. 14: 131-6
9. Cai J, Chen J, Zhang W, Yang P, Zhang C, Li M. Loss of ATRX, associated with DNA methylation pattern of chromosome end, impacted biological behaviors of astrocytic tumors. Oncotarget. 2015. 6: 18105-15
10. Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998. 90: 1473-9
11. Cancer Genome Atlas Research Netwo. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008. 455: 1061-8
12. Dittmann K, Mayer C, Rodemann HP. Inhibition of radiation-induced EGFR nuclear import by C225 (Cetuximab) suppresses DNA-PK activity. Radiother Oncol. 2005. 76: 157-61
13. Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med. 2015. 372: 2499-508
14. Griffin CA, Burger P, Morsberger L, Yonescu R, Swierczynski S, Weingart JD. Identification of der(1;19)(q10;p10) in five oligodendrogliomas suggests mechanism of concurrent 1p and 19q loss. J Neuropathol Exp Neurol. 2006. 65: 988-94
15. Guo CF, Zhuang Y, Chen Y, Chen S, Peng H, Zhou S. Significance of tumor protein p53 mutation in cellular process and drug selection in brain lower grade (WHO grades II and III) glioma. Biomark Med. 2020. 14: 1139-50
16. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022. 12: 31-46
17. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000. 100: 57-70
18. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011. 144: 646-74
19. Huang LE. Impact of CDKN2A/B homozygous deletion on the prognosis and biology of IDH-mutant glioma. Biomedicines. 2022. 10: 246
20. Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006. 66: 9852-61
21. Killela PJ, Pirozzi CJ, Healy P, Reitman ZJ, Lipp E, Rasheed BA. Mutations in IDH1, IDH2, and in the TERT promoter define clinically distinct subgroups of adult malignant gliomas. Oncotarget. 2014. 5: 1515-25
22. Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013. 110: 6021-6
23. Koschmann C, Lowenstein PR, Castro MG. ATRX mutations and glioblastoma: Impaired DNA damage repair, alternative lengthening of telomeres, and genetic instability. Mol Cell Oncol. 2016. 3: e1167158
24. Leeper HE, Caron AA, Decker PA, Jenkins RB, Lachance DH, Giannini C. IDH mutation, 1p19q codeletion and ATRX loss in WHO grade II gliomas. Oncotarget. 2015. 6: 30295-305
25. Lopez-Gines C, Cerda-Nicolas M, Gil-Benso R, Pellin A, Lopez-Guerrero JA, Callaghan R. Association of chromosome 7, chromosome 10 and EGFR gene amplification in glioblastoma multiforme. Clin Neuropathol. 2005. 24: 209-18
26. Lopez-Gines C, Gil-Benso R, Ferrer-Luna R, Benito R, Serna E, Gonzalez-Darder J. New pattern of EGFR amplification in glioblastoma and the relationship of gene copy number with gene expression profile. Mod Pathol. 2010. 23: 856-65
27. Louis DN, Aldape K, Brat DJ, Capper D, Ellison DW, Hawkins C. Announcing cIMPACT-NOW: The consortium to inform molecular and practical approaches to CNS tumor taxonomy. Acta Neuropathol. 2017. 133: 1-3
28. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007. 114: 97-109
29. Louis DN, Perry A, Reifenberger G, von Deimling A, FigarellaBranger D, Cavenee WK. The 2016 world health organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016. 131: 803-20
30. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, FigarellaBranger D. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro Oncol. 2021. 23: 1231-51
31. Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: Progress and puzzles. Curr Opin Genet Dev. 2003. 13: 77-83
32. Marker DF, Agnihotri S, Amankulor N, Murdoch GH, Pearce TM. The dominant TP53 hotspot mutation in IDH-mutant astrocytoma, R273C, has distinctive pathologic features and sex-specific prognostic implications. Neurooncol Adv. 2022. 4: vdab182
33. Mizoguchi M, Betensky RA, Batchelor TT, Bernay DC, Louis DN, Nutt CL. Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: Correlation with EGFR status, tumor grade, and survival. J Neuropathol Exp Neurol. 2006. 65: 1181-8
34. Morin GB. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell. 1989. 59: 521-9
35. Mukherjee B, McEllin B, Camacho CV, Tomimatsu N, Sirasanagandala S, Nannepaga S. EGFRvIII and DNA double-strand break repair: A molecular mechanism for radioresistance in glioblastoma. Cancer Res. 2009. 69: 4252-9
36. Nandakumar P, Mansouri A, Das S. The role of ATRX in glioma biology. Front Oncol. 2017. 7: 236
37. Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro Oncol. 2014. 16: iv1-63
38. Pekmezci M, Rice T, Molinaro AM, Walsh KM, Decker PA, Hansen H. Adult infiltrating gliomas with WHO 2016 integrated diagnosis: Additional prognostic roles of ATRX and TERT. Acta Neuropathol. 2017. 133: 1001-16
39. Pinkham MB, Telford N, Whitfield GA, Colaco RJ, O’Neill F, McBain CA. FISHing tips: What every clinician should know about 1p19q analysis in gliomas using fluorescence in situ hybridisation. Clin Oncol (R Coll Radiol). 2015. 27: 445-53
40. Pirozzi CJ, Yan H. The implications of IDH mutations for cancer development and therapy. Nat Rev Clin Oncol. 2021. 18: 645-61
41. Rasheed BK, McLendon RE, Herndon JE, Friedman HS, Friedman AH, Bigner DD. Alterations of the TP53 gene in human gliomas. Cancer Res. 1994. 54: 1324-30
42. Reifenberger J, Reifenberger G, Liu L, James CD, Wechsler W, Collins VP. Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol. 1994. 145: 1175-90
43. Reis GF, Pekmezci M, Hansen HM, Rice T, Marshall RE, Molinaro AM. CDKN2A loss is associated with shortened overall survival in lower-grade (World Health Organization Grades II-III) astrocytomas. J Neuropathol Exp Neurol. 2015. 74: 442-52
44. Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J Natl Cancer Inst. 2010. 102: 932-41
45. Roth JJ, Fierst TM, Waanders AJ, Yimei L, Biegel JA, Santi M. Whole chromosome 7 gain predicts higher risk of recurrence in pediatric pilocytic astrocytomas independently from KIAA1549-BRAF fusion status. J Neuropathol Exp Neurol. 2016. 75: 306-15
46. Saadeh FS, Mahfouz R, Assi HI. EGFR as a clinical marker in glioblastomas and other gliomas. Int J Biol Markers. 2018. 33: 22-32
47. Sahm F, Reuss D, Koelsche C, Capper D, Schittenhelm J, Heim S. Farewell to oligoastrocytoma: In situ molecular genetics favor classification as either oligodendroglioma or astrocytoma. Acta Neuropathol. 2014. 128: 551-9
48. Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio II. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012. 26: 2038-49
49. Shalaby T, Hiyama E, Grotzer MA. Telomere maintenance as therapeutic target in embryonal tumours. Anticancer Agents Med Chem. 2010. 10: 196-212
50. Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003. 63: 6962-70
51. Shirahata M, Ono T, Stichel D, Schrimpf D, Reuss DE, Sahm F. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 2018. 136: 153-66
52. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012. 483: 479-83
53. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010. 17: 98-110
54. Wang H, Cai S, Bailey BJ, Reza Saadatzadeh M, Ding J, Tonsing-Carter E. Combination therapy in a xenograft model of glioblastoma: Enhancement of the antitumor activity of temozolomide by an MDM2 antagonist. J Neurosurg. 2017. 126: 446-59
55. Weller M, van den Bent M, Preusser M, Le Rhun E, Tonn JC, Minniti G. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol. 2021. 18: 170-86
56. Weller M, Wick W, Aldape K, Brada M, Berger M, Pfister SM. Glioma. Nat Rev Dis Primers. 2015. 1: 15017
57. Wiestler B, Capper D, Holland-Letz T, Korshunov A, von Deimling A, Pfister SM. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol. 2013. 126: 443-51
58. Wu N, Lin X, Zhao X, Zheng L, Xiao L, Liu J. MiR-125b acts as an oncogene in glioblastoma cells and inhibits cell apoptosis through p53 and p38MAPK-independent pathways. Br J Cancer. 2013. 109: 2853-63
59. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009. 360: 765-73

